Novinky v léčbě primární biliární cholangitidy
Libuše Husová Orcid.org 1
+ Pracoviště
Souhrn
Primární biliární cholangitida je chronické, imunologicky podmíněné, postupně progredující jaterní onemocnění charakterizované destrukcí malých žlučovodů, jejímž důsledkem je cholestáza, jaterní fibróza s rozvojem jaterní cirhózy se všemi komplikacemi. Ty pak v konečné fázi vedou k selhání jater s následkem smrti nebo potřeby transplantace jater. Včasná a účinná terapie je klíčovým faktorem ovlivnění prognózy. Onemocnění postihuje převážně ženy, a to v 90 %. Diagnóza je možná na základě elevace sérové alkalické fosfatázy (ALP) nad 1,5násobek normy po dobu 6 měsíců a v kombinaci s přítomností antimitochondriálních protilátek (AMA) ≥ 1: 40, nebo specifických antinukleárních protilátek (ANA) sp100, gp210, nebo kombinace zvýšené sérové ALP s typickým histologickým nálezem v jaterní biopsii. Přestože kyselina ursodeoxycholová (UDCA) zůstává léčbou první volby, někteří pacienti tuto léčbu netolerují nebo odpověď na tuto léčbu není dostatečná. V současné době je již dostupná léčba 2. linie – agonisté receptorů aktivovaných proliferátory peroxisomů (PPAR), delta (PPAR-δ) či duální agnonisté (PPAR-α/δ). Seladelpar (PPAR-δ) a elafibranor (PPAR-α/δ) jsou slibnou alternativou léčby pro své protizánětlivé a antifibrotické vlastnosti.
Klíčová slova
alkalická fosfatáza, primární biliární cholangitida, AMA protilátky, ANA protilátky, kyselina ursodeoxycholová, elafibranor, seladelpar
Úvod
Primární biliární cholangitida (PBC) je chronické, imunologicky podmíněné, progredující jaterní onemocnění charakterizované progresivní destrukcí malých nitrojaterních žlučovodů, jejímž důsledkem je cholestáza, progresivní jaterní fibróza, která vede v konečné fázi až k selhání jater s následkem smrti nebo potřeby transplantace jater [1,2]. Choroba byla poprvé popsána Addisonem a Gullem v roce 1851 [3]. Později byla přejmenována na „xantomatózní biliární cirhózu“ podle charakteristických xantomů na kůži a přítomnosti biliární cirózy [4]. Pojmenování primární biliární cirhóza bylo používáno od roku 1950 a vyjadřovalo přítomnost klinických projevů této choroby, neboť choroba byla diagnostikována ve stadiu rozvinuté jaterní cirhózy [5]. U některých pacientů nebyla v době diagnózy přítomna rozvinutá jaterní cirhóza [1], a tak bylo jasné, že název není zcela správný. To vedlo v roce 2014 ke změně pojmenování s cílem odstranit stigma cirhózy, špatné prognózy a spojení s etiologií cirhózy s alkoholem. Změna pojmenování na primární biliární cholangitidu byla schválena Evropskou asociací pro studium jater (EASL) v roce 2014 a Americkou asociací pro studium jater (AASLD) v roce 2015 [6,7]. V současnosti nemáme data pro incidenci a prevalenci PBC v ČR. V Evropě je incidence udávána v rozmezí 0,3–5,8 nemocných na 100 000 a rok, prevalence 1,9–40 na 100 000 [7]. Onemocnění postihuje především ženy (10: 1 k mužům) mladšího a středního věku. Relativním rizikovým faktorem je rodinná anamnéza PBC v první linii. Dalším nezávislým rizikovým faktorem je přítomnost jiného autoimunního onemocnění (Sjögrénův sybdrom, autoimunní thyreoiditida, psoriáza, revmatoidní artritida a další) [8]. Etiologie a patofyziologie není dostatečně objasněna. V současné době je PBC považována za orgánově specifické autoimunní onemocnění, které postihuje geneticky disponované jedince [7].
Klinické projevy
Většina pacientů nemá žádné příznaky, je tedy klinicky asymptomatická. Na diagnózu PBC je nutné myslet na základě náhodného nálezu cholestázy, zejména elevace ALP. Pro klinickou manifestaci jsou typické příznaky únavy a pruritu, které často předchází diagnostiku o mnoho let. U některých nemocných je přítomna bolest v pravém podžebří [7]. Nejčastějším klinickým příznakem je únava, kterou udává 85 % nemocných, je také příčinou zhoršené kvality života těchto nemocných [7,9]. Dalším častým příznakem je pruritus, který je definován jako nepříjemný pocit nutící nemocného ke škrábání, vyskytuje se u 70 % pacientů s PBC [7,10]. Pruritus postihuje kůži celého těla a rovněž výrazně snižuje kvalitu života, způsobuje nespavost a depresivní ladění pacienta. Klinicky manifestní se choroba stává s rozvojem jaterní cirhózy, a to nejčastěji ve fázi dekompenzace, jak vaskulární, tak metabolické. Choroba je rovněž charakteristická možnými extrahepatálními příznaky, mezi ně patří osteoporóza, hyperlipidemie či přítomnost jiného autoimunního onemocnění. Osteoporóza bývá u 20–40 % pacientů s PBC a zvyšuje riziko vzniku fraktur. Riziko osteoporózy zvyšuje vyšší věk, nízký BMI a pokročilé stadium choroby. Cholestáza vede k malabsorbci vitaminů rozpustných v tucích (A, D, E, K), a tím ke zvýšení rizika osteoporózy [11]. Hyperlipidemie provází až 85 % pacientů s PBC. Častým projevem je přítomnost xantelazmat a xantomů na kůži pacientů. Souvislost mezi hyperlipidemií a kardiovaskulárním onemocněním nebyla doposud prokázána [12].
Diagnóza
Diagnóza je stanovena na základě kombinace přítomnosti cholestázy, elevace ALP > 1,5násobek horní hranice normy trvající > 6 měsíců a přítomnosti AMA protilátek v titru 1: 40 nebo vyšším. U AMA negativních jedinců s přítomnou cholestázou mohou být pozitivní specifické ANA protilátky (sp100, gp210), které v tomto případě mohou potvrdit diagnózu PBC při současné cholestáze. V případě negativity AMA i specifických ANA protilátek u pacientů, u nichž je přítomna cholestáza, je pak nutné provedení jaterní biopsie s histologickou verifikací PBC. U pacientů s AMA pozitivitou bez cholestázy je riziko rozvoje PBC 14–15 % do 5 let. Tuto skupinu pacientů je vhodné dispenzarizovat v intervalech 1krát ročně [7]. V histologickém nálezu je charakteristická přítomnost nehnisavé cholangitidy s destrukcí malých interlobulárních a septálních žlučovodů. Zánětlivá lymfocytární infiltrace v okolí malých žlučovodů je tvořena převážně T-lymfocyty a v menším množství B-lymfocyty, makrofágy a eozinofily. Typická je přítomnost epiteloidních granulomů v blízkosti žlučovodů. Z uvedeného vyplývá, že jaterní biopsie není vždy nutnou podmínkou pro stanovení správné diagnózy [7]. Typickým nálezem u PBC je cholestáza, dominance ALP a GGT (-glutamyl transferáza), hodnoty AST (asparátaminotrasferáza) a ALT (alaninaminotransferáza) mohou být rovněž zvýšeny. Při elevaci AST a ALT musíme myslet na možnost překryvného syndromu s autoimunní hepatitidou (AIH). V laboratorním obraze je charakteristická hyperlipidemie a přítomnost elevace imunoglobulinů ve třídě IgM. U pokročilé PBC ve stadiu jaterní cirhózy může být zvýšený bilirubin, pokles albuminu, trombocytů a prodloužený protrombinový čas. Základem diagnostiky je u přítomné cholestázy provedení ultrazvukového vyšetření, které nám vyloučí obstrukci, ale zároveň umožní zhodnocení struktury jater, přítomnost ascitu, portální hypertenze, velikost sleziny nebo přítomnost ložiskového procesu jater [2,7]. Prognózu pacientů s PBC zhoršuje přítomnost jaterní cirhózy. V současné době je většina pacientů diagnostikována v časných fázích onemocnění a díky léčbě nedospějí do terminálního stadia jaterní choroby. Pravděpodobně nejlepším prognostickým markrem je hodnota ALP v séru, která dle multivariantní analýzy téměř 5 000 pacientů s PBC koreluje s rizikem úmrtí či potřebou transplantace jater [13].
Léčba
Kauzální terapie PBC není známá. Dostupná terapie je zaměřena na zpomalení progrese PBC do konečných stadií jaterního onemocnění a neméně důležitá je terapie klinických projevů a komplikací (únavy, pruritu, osteoporózy) a v případě rozvinuté jaterní cirhózy pak terapie všech jejích komplikací. V posledních letech se do klinické praxe dostávají nové preparáty, které lze využít v terapii PBC, a tím zlepšit prognózu těchto pacientů. Léčba je indikována v případě nedostatečné odpovědi na léčbu první linie kyselinou ursodeoxycholovou (UDCA) nebo při její intoleranci. Ve druhé linii léčby jsou v současné době v České republice užívány dva preparáty, a to elafibranor a seladelpar.
Léčba první linie
Kyselina ursodeoxycholová
Na základě klinických studií je považována za lék první linie k léčbě PBC. V klinické praxi byl efekt UDCA na průběh PBC pozitivně testován v různých dávkách (5–25 mg/kg/den). Za optimální je považována dávka 13–15 mg/kg/den, ta byla potvrzena v několika studiích a v následné metaanalýze sedmi randomizovaných studií s více než 1 000 pacienty z roku 2006. Tato metaanalýza ukázala snížení úmrtí i potřeby transplantace jater u pacientů léčených uvedenou dávkou [14,15]. Studie Global C z roku 2014, která hodnotila 4 845 nemocných v horizontu 5, 10 a 15 let v porovnání s neléčenou skupinou, prokázala signifikantní rozdíl (90 vs. 78 %, 66 vs. 79 % a 59 vs. 32 %) v prodloužení intervalu do doby transplantace i přežívání [13,16]. Léčba UDCA je považována za bezpečnou, s minimem nežádoucích účinků. Jde o léčbu, kterou je možné užít i v těhotenství a po porodu [7]. Vzhledem k faktu, že pokles ALP u pacientů léčených UDCA zlepšuje jejich prognózu, prodlužuje dobu do transplantace a zlepšuje přežívání, byly vytvořeny prognostické modely k predikci prognózy pacientů s PBC. Patří mezi ně „Barcelonská kritéria“ – pokles ALP k normě nebo > 40 % po 12 měsících léčby, „Pařížská kritéria“ – hodnoty bilirubinu ≤ 17 μmol/l, ALP ≤ 3násobek normálních hodnot (ULN) a AST ≤ 2násobek normy rovněž po roce léčby UDCA, „Torontská kritéria“ – pokles ALP ≤ 1,67násobek normy po 12 a 24 měsících. Nejužívanějším kritériem účinnosti léčby je pokles ALP ≤ 1,67násobek normy po 12 měsících terapie [7,16–19]. Jednoznačně nejlepší přežití bez komplikací mají pacienti, kteří dosáhnou normalizace ALP (tzv. hluboká odpověď). To prokazuje i studie z 24 center u 1 047 pacientů. Ti, kteří dosáhli normalizace ALP, měli signifikantně nižší riziko komplikací ve srovnání s pacienty, kteří nedosáhli normalizace ALP, ale na léčbu odpověděli (ALP < 1,67 × ULN), a to včetně pacientů s tranzientní elastografií ≥ 10,0 kPa [20]. Existují však pacienti, kteří na léčbu UDCA neodpoví nebo ji netolerují, pro tuto skupinu pacientů je v současné době dostupná léčba druhé linie.
Léčba druhé linie
V současné době jsou v České republice dostupné dva klinicky ověřené preparáty: elafibranor a seladelpar.
Elafibranor
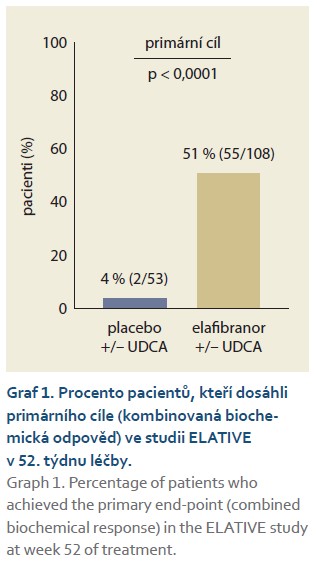
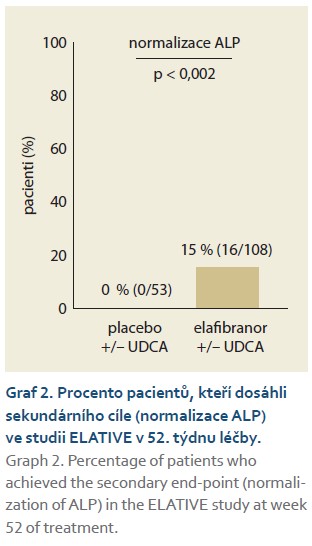
Elafibranor a jeho hlavní aktivní metabolit GFT1007 jsou duální agonisté receptorů aktivovaných proliferátory peroxizomů (PPAR-α/δ). Má se za to, že PPAR-α/δ jsou klíčovými regulátory homeostázy žlučových kyselin, zánětu a fibrózy. Aktivace PPAR-α a PPAR-δ snižuje toxicitu žluči a zlepšuje cholestázu modulováním syntézy, detoxifikace a transportérů žlučových kyselin. Aktivace PPAR-α a PPAR-δ má rovněž protizánětlivé účinky [21,22]. Účinnost elafibranoru byla hodnocena ve studii GFT505B-319-1 (ELATIVE), randomizované, dvojitě zaslepené, placebem kontrolované studii fáze 3 u 161 dospělých s PBC s nedostatečnou odpovědí nebo intolerancí UDCA. Hodnocení léčby bylo po dobu 52 týdnů při podávání 80 mg elafibranoru 1krát denně. Do studie nebyli zahrnuti pacienti s dekompenzovanou jaterní cirhózou. Primárním cílovým parametrem byla odpověď cholestázy v 52. týdnu definovaná jako složený cílový parametr: ALP < 1,67 × ULN a celkový bilirubin ≤ ULN a snížení ALP ≥ 15 % (graf 1). Sekundárními cílovými parametry byly normalizace ALP v 52. týdnu (graf 2). Dalším sekundárním cílem byla změna pruritu na základě skóre WI-NRS priritu (Worst Itch Numeric Rating Scale – číselné hodnoticí škály pruritu) u nemocných se středně těžkým až těžkým pruritem v době zařazení do studie. Pokles pruritu nebyl v této studii statisticky významný [22,23]. Elafibranor se podává v dávce 80 mg jednou denně v kombinaci s UDCA u dospělých pacientů s nedostatečnou odpovědí na UDCA. U nemocných, kteří terapii UDCA netolerují, se podává elafibranor v monoterapii. Při léčbě elafibranorem se může vyskytnout zvýšení kreatinkinázy, proto je vhodné její hladinu stanovit před zahájením léčby. Jako nejčastější nežádoucí účinky se ve studii ELATIVE vyskytly bolesti břicha, průjem, nauzea a zvracení. Jednalo se o nezávažné, mírné až středně těžké nežádoucí účinky, které se vyskytly v časném stadiu léčby a obvykle odezněly v průběhu dnů až několika týdnů bez jakékoli úpravy dávky nebo podpůrných opatření [22]. U starších pacientů není nutná úprava dávky, ta rovněž není nutná u pacientů s poruchou funkce ledvin. Elafibranor lze rovněž podávat bez úpravy dávky u pacientů s jaterní cirhózou s lehkou a střední jaterní dysfunkcí (CHP A, B), u pacientů s těžkou jaterní dysfunkcí (CHP C) se podání elafibranoru nedoporučuje [23].
Seladelpar
Dalším lékem, který lze užít v terapii PBC při nedostatečné odpovědi nebo intoleranci UDCA, je seladelpar. Seladelpar je agonista receptorů aktivovaných proliferátory PPAR-δ neboli delparu. PPAR-δ je jaderný receptor exprimovaný v játrech a dalších tkáních. Aktivace PPAR-δ snižuje syntézu žlučových kyselin v játrech prostřednictvím down regulace CYP7A1, klíčového enzymu pro syntézu žlučových kyselin z cholesterolu, závislé na fibroblastovém růstovém faktoru 21 (FGF21), a snížením syntézy a absorpce cholesterolu. Tyto účinky vedou k nižší expozici jater žlučovým kyselinám a k poklesu hladin cirkulujících žlučových kyselin. Účinnost seladelparu byla hodnocena u pacientů s PBC v randomizované, dvojitě zaslepené, placebem kontrolované 12měsíční studii (RESPONSE). Do studie byli zařazeni pacienti, u nichž hodnota koncentrace ALP byla ≥ 1,67násobek horní hranice normálních hodnot a koncentrace celkového bilirubinu byla ≤ 2 × ULN. Ze studie byli vyloučeni pacienti s jinými chronickými jaterními onemocněními nebo s klinicky významnou jaterní dekompenzací (metabolickou i vaskulární, CHP C). Čtrnáctidenní přípravné období před randomizací sloužilo ke stanovení výchozí intenzity svědění, která byla měřena pacientem během 24 hod pomocí číselné hodnoticí škály pruritu (WI-NRS pruritu) (skóre 0 = „žádné svědění“ až 10 = „nejhorší možné svědění“). Pacienti byli randomizováni (2: 1) k podávání seladelparu (n = 128) v dávce 10 mg jednou denně, nebo placeba (n = 65) po dobu 12 měsíců. Seladelpar nebo placebo byly během studie podávány v kombinaci s UDCA u 181 (94 %) pacientů, nebo jako monoterapie u 12 (6 %) pacientů s intolerancí UDCA.
Obě skupiny pacientů byly obecně vyvážené z hlediska výchozích demografických údajů a charakteristik onemocnění. Ve skupině 193 pacientů byl věkový průměr 56,7 roku (rozmezí 28–75 let); 41 pacientů (21 %) bylo ve věku 65 let nebo více; 183 (95 %) byly ženy.
Průměrná výchozí koncentrace ALP byla 314,3 U/l, což odpovídalo 2,7násobku ULN. Průměrná výchozí koncentrace celkového bilirubinu byla 0,758 mg/dl a byla nižší nebo rovna ULN u 87 % zařazených pacientů. Středně těžký až těžký pruritus byl hodnocen dle skóre WI-NRS ≥ 4. Středně těžký nebo těžký pruritus mělo na počátku studie 38 % pacientů ve větvi se seladelparem (WI-NRS = 6,1) a 35 % pacientů s placebem (WI-NRS = 6,6). Cirhózu (CHP A) mělo na počátku studie 18 pacientů (14 %) v rameni se seladelparem 10 mg a 9 pacientů (14 %) v rameni s placebem.
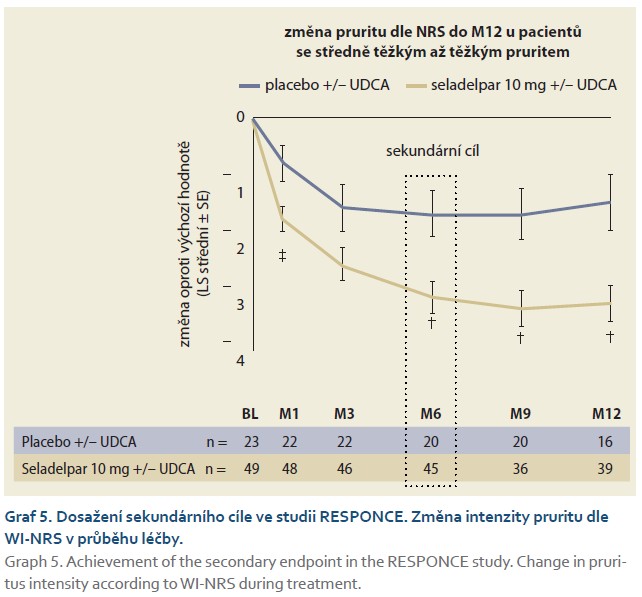
Ve studii RESPONSE byla primárním cílovým parametrem analýza odpovědí ve 12. měsíci, kde odpověď byla definována jako kombinace tří kritérií:
- koncentrace ALP < 1,67násobek ULN;
- koncentrace celkového bilirubinu ≤ ULN;
- pokles koncentrace ALP o alespoň 15 %.
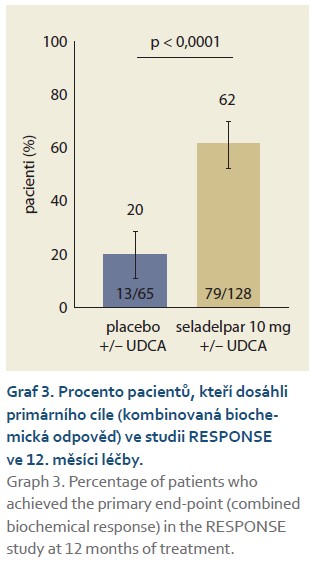
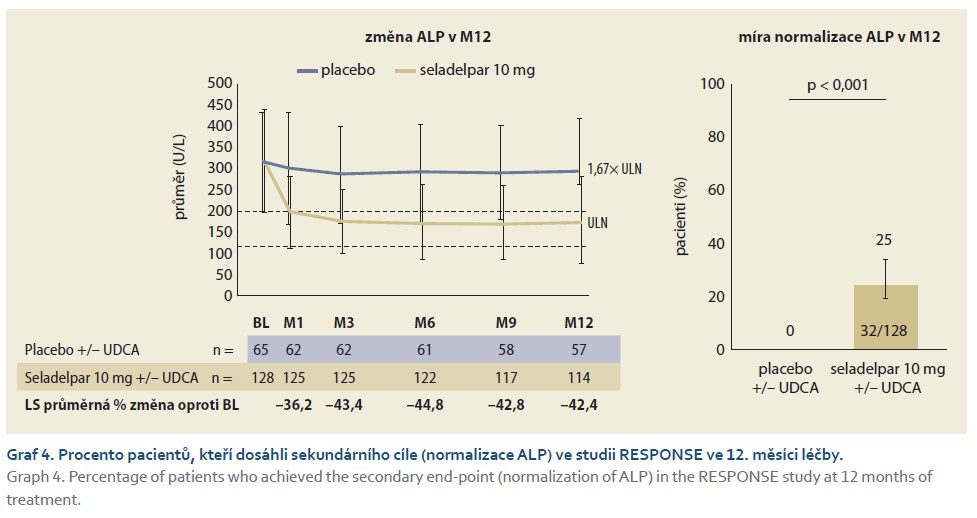
Normalizace hladiny ALP byla definována jako dosažení koncentrace ALP ≤ ULN. Zlepšení pruritu bylo hodnoceno podle změny oproti výchozí hodnotě v týdenním průměru skóre WI-NRS pruritu v 6. měsíci u pacientů se skóre WI-NRS pruritu ≥ 4 na počátku studie. Pacienti léčení seladelparem statisticky významně dosáhli primárního cíle oproti pacientům s placebem (graf 3). A rovněž normalizace ALP (sekundární cíl) byla významně vyšší u pacientů léčených seladelparem oproti placebu ve 12. měsíci léčby (graf 4). Poklesy ALP byly pozorovány v 1. měsíci, pokračovaly do 6. měsíce a přetrvávaly až do 12. měsíce. Seladelpar signifikantně snížil pruritus ve srovnání s placebem v 6. měsíci u pacientů s průměrným výchozím skóre pruritu ≥ 4 podle WI-NRS pruritu, což je klíčový sekundární cílový parametr ve studii RESPONSE. Seladelpar vedl ke snížení intenzity pruritu hlášené pacienty v 1. měsíci, přičemž toto snižování pokračovalo až do 6. měsíce (p < 0,005). Signifikantní snížení pruritu (sekundární cíl) u pacientů léčených seladelparem je dokumentováno v grafu 5. U starších pacientů není nutná úprava dávky, ta rovněž není nutná u pacientů s poruchou funkce ledvin [24,25].
V metaanalýze tří randomizovaných studií o 496 pacientech seladelpar významně zlepšil normalizaci ALP ve srovnání s placebem, a navíc snížil hodnoty ALP po zahájení léčby u pacientů s PBC. Z nežádoucích účinků se nejčastěji vyskytly bolesti břicha a hlavy [26]. Přípravek seladelpar je indikován k léčbě PBC v kombinaci s kyselinou ursodeoxycholovou u dospělých pacientů, kteří mají nedostatečnou odpověď na samotnou UDCA, nebo jako monoterapie u dospělých pacientů s intolerancí UDCA [24,25]. Přestože prokázal terapeutické přínosy, je třeba provést další výzkum k vyhodnocení jeho dlouhodobé bezpečnosti, zejména pokud jde o výskyt nežádoucích účinků, a k určení jeho účinnosti při různých dávkách [26].
Závěr
Primární biliární cholangitida je chronické autoimunní onemocnění, které je charakterizováno cholestázou trvající déle než 6 měsíců. Diagnóza je založena na přítomnosti cholestázy, průkazu specifických protilátek a/nebo typickém histologickém nálezu. Neléčené onemocnění vede k rozvoji jaterní cirhózy s jejími možnými komplikacemi, které vedou k transplantaci jater či smrti pacienta. Bohužel kauzální léčba není známá, cílem léčby je oddálení vzniku cirhózy a jejích komplikací. Z tohoto důvodu je naším cílem diagnostikovat PBC před rozvojem jaterní cirhózy a včasnou léčbou oddálit progresi tohoto onemocnění. Lékem první volby je UDCA. V případě netolerance nebo nedostatečné odpovědi na léčbu UDCA je v současné době možná léčba druhé linie: elafibranor, seladerpar. Současně je nutná léčba symptomů této choroby, a to zejména pruritu a kostní choroby. Seladelpar signifikantně snižuje pruritus u pacientů se středně těžkým nebo těžkým pruritem, a proto je pro tyto pacienty výhodnější. U nemocných s dekompenzovanou jaterní cirhózou při PBC je pak nutné včas indikovat transplantaci jater, která je jistě léčebnou a život zachraňující metodou.
ORCID autorky
L. Husová 0000-0002-9639-2541.
Doručeno/Submitted: 27. 8. 2025
Přijato/Accepted: 11. 9. 2025
MUDr. Libuše Husová, Ph.D.
Centrum kardiovaskulární a transplantační chirurgie
Pekařská 53
602 00 Brno
libuse.husova@cktch.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Sherlock S. Primary billiary cirrhosis (chronic intrahepatic obstructive jaundice). Gastroenterology 1959; 37: 574–586.
2. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 2009; 51(2): 237–267. doi: 10.1016/j.jhep.2009.04.009.
3. Addison T, Gull W. On a certain affection of the skin-vitiligoidea-a plana, B tuberosa. Guys Hosp Rep 1851; 7: 265–276.
4. Mac MH, Thannhauser SJ. Xanthomatous biliary cirrhosis; a clinical syndrome. Ann Intern Med 1949; 30(1): 121–179. doi: 10.7326/0003-4819-30-1-121.
5. Ahrens EH, Payne MA, Kunkel HG et al. Primary biliary cirrhosis. Medicine 1950; 29(4): 299–364. doi: 10.1097/00005792-195012000-00002.
6. Beuers U, Gershwin ME, Gish RG et al. Changing nomenclature for PBC: from „cirrhosis“ to „cholangitis“. J Hepatol 2015; 63(5): 1285–1287. doi: 10.1016/j.jhep.2015.06.031.
7. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017; 67(1): 145–172. doi: 10.1016/j.j hep.2017.03.022.
8. Gershwin ME, Selmi C, Worman HJ et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology 2005; 42(5): 1194–1202. doi: 10.1002/hep.20907.
9. Jones DE, Bhala N, Burt J et al. Four year follow-up of fatigue in a geographically defined primary biliary cirrhosis patient cohort. Gut 2006; 55(4): 536–541. doi: 10.1136/gut.2005.080317.
10. Prince M, Chetwynd A, Newman W et al. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years. Gastroenterology 2002; 123(4): 1044–1051. doi: 10.1053/gast.2002.36027.
11. Phillips JR, Angulo P, Petterson T et al. Fat-soluble vitamin levels in patients with primary biliary cirrhosis. Am J Gastroenterol 2001; 96(9): 2745–2750. doi: 10.1111/j.1572-0241.2001.04134.x.
12. Sorokin A, Brown JL, Thompson PD. Primary biliary cirrhosis, hyperlipidemia, and atheroscierotic risk: a systematic review. Atherosclerosis 2007; 194(2): 293–299. doi: 10.1016/j.atherosclerosis.2006.11.036.
13. Lammers WJ, van Buuren HR, Hirschfield GM et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology 2014; 147(6): 1338.e5–1349.e5. doi: 10.1053/ j.gastro.2014.08.029.
14. Shi J, Wu C, Lin Y et al. Long-term effects of mid-dose ursodeoxycholic acid in primary biliary cirrhosis: a meta-analysis of randomized controlled trials. Am J Gastroenterol 2006; 101(7): 1529–1538. doi: 10.1111/j.1572-0241.2006.00634.x.
15. Corpechot C, Carrat F, Bonnand AM et al. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology 2000; 32(6): 1196–1199. doi: 10.1053/jhep.2000.20240.
16. Lammers WJ, Hirschfield GM, Corpechot C et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology 2015; 149(7): 1804.e4–1812.e4. doi: 10.1053/ j.gastro.2015.07.061.
17. Murillo Perez CF, Harms MH, Lindor KD et al. Goals of treatment for improved survival in primary biliary cholangitis: treatment target should be bilirubin within the normal range and normalization of alkaline phosphatase. Am J Gastroenterol 2020; 115(7): 1066–1074. doi: 10.14309/ajg.0000000000000557.
18. Kumagi T, Guindi M, Fischer SE et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol 2010; 105(10): 2186–2194. doi: 10.1038/ ajg.2010.216.
19. Nevens F, Andreone P, Mazzella G et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016; 375(7): 631–643. doi: 10.1056/NEJMoa1509840.
20. Corpechot C, Lemoinne S, Soret PA et al. Adequate versus deep response to ursodeoxycholic acid in primary biliary cholangitis: to what extent and under what conditions is normal alkaline phosphatase level associated with complication-free survival gain? Hepatology 2024; 79(1): 39–48. doi: 10.1097/ HEP.0000000000000529.
21. Colapietro F, Gershwin ME, Lleo A. PPAR agonists for the treatment of primary biliary cholangitis: old and new tales. J Transl Autoimmun 2023; 6: 100188. doi: 10.1016/j.jtauto.2023.100188.
22. SPC Iqirvo 2025. Souhrn údajů o přípravku. 2025 [online]. Dostupné z: chrome-extension: // efaidnbmnnnibpcajpcglclefindmkaj/https: //ec.europa.eu/health/documents/community-register/2024/20240919163733/anx_163733_ cs.pdf.
23. Kowdley KV, Bowlus CL, Levy C et al. Efficacy and safety of elafibranor in primary biliary cholangitis. N Engl J Med 2024; 390(9): 795–805. doi: 10.1056/ NEJMoa2306185.
24. SPC Lyvdelzi. Souhrn údajů o přípravku. 2025 [online]. Dostupné z: chrome-extension: //efaidnbmnnnibpcajpcglclefindmkaj/ https: //www.ema.europa.eu/cs/documents/ product-information/lyvdelzi-epar-product-information_cs.pdf.
25. Hierschfield GM, Bowlus CL, Mayo MJ et al. A phase 3 trial of seladelpar in primary biliary cholangitis. N Engl J Med 2024; 390(9): 783–794. doi: 10.1056/NEJMoa2312100.
26. Ashraf T, Abunada O, Seerani N et al. The role of seladelpar in primary biliary cholangitis: a systematic review and meta-analysis. BMC Gastroenterology 2025; 25: 224. doi: 10.1186/s12876-025-03812-3.