Význam genetického vyšetrenia u detí s idiopatickou chronickou pankreatitídou
Ester Hegyi1, Ľudmila Vávrová2, Michal Konečný Orcid.org 3, László Kovács Orcid.org 1, Iveta Čierna1
+ Pracoviště
Souhrn
Zatiaľčo u dospelých sa ako hlavný etiologický faktor chronickej pankreatitídy uvádza nadmerná konzumácia alkoholu, u detí sa okrem anatomických anomálií pankreasu a žlčových ciest uplatňujú predovšetkým genetické faktory. Cieľ: Charakterizovať frekvenciu výskytu mutácií jednotlivých génov asociovaných so vznikom rekurentnej akútnej pankreatitídy alebo chronickej pankreatitídy u detí a následne sledovať genotypovo-fenotypové korelácie u týchto pacientov. Materiál a metodika: Do štúdie bolo zaradených 21 detí s diagnózou rekurentnej akútnej pankreatitídy/chronickej pankreatitídy nejasnej etiológie, ktoré boli v období 2008–2013 vyšetrené na II. detskej klinike LF UK a DFNsP v Bratislave. Molekulovo-genetická analýza génov PRSS1, SPINK1 a CTRC sa uskutočnila v spolupráci s Oddelením lekárskej genetiky Onkologického ústavu sv. Alžbety. Výsledky: Rodinná anamnéza bola pozitívna v piatich prípadoch. U dvoch pacientov bola dokázaná prítomnosť mutácie p.R122H génu PRSS1; jeden pacient je zmiešaný heterozygot pre mutácie p.G208A (PRSS1) a p.G60G (CTRC). Mutácia p.N34S génu SPINK1 sa potvrdila u šiestich pacientov (dvaja homozygotní a štyria heterozygotní), z nich u štyroch bola potvrdená aj mutácia p.G60G (CTRC) v heterozygotnom stave. Jeden pacient je homozygot pre mutáciu p.G60G; jeden heterozygot pre p.R254W a jeden heterozygot pre mutáciu p.G214R génu CTRC. Záver: V súbore 21 detských pacientov s rekurentnou akútnou pankreatitídou/chronickou pankreatitídou neznámej etiológie sme zaznamenali vysoký výskyt kauzálnych mutácií asociovaných so vznikom ochorenia. Uvedené výsledky potvrdzujú dôležitosť genetického vyšetrenia u detí s idiopatickou rekurentnou akútnou pankreatitídou /chronickou pankreatitídou.
Klíčová slova
chronická pankreatitida, genetická analýza, genetická náchylnost, hereditárna pankreatitída, pankreatitída u detíÚvod
Chronická pankreatitída (CHP) patrí medzi zriedkavé ochorenia detského veku. Jej incidencia sa vo vekovej skupine mladších ako 20 rokov odhaduje na 0,5/100 000, vekom však postupne narastá a v dospelosti sa vyznačuje až 4–9× vyšším výskytom v porovnaní s detskou populáciou [1]. Ochorenie je definované ako trvalo progredujúci zápalový proces podžalúdkovej žľazy vedúci k vzniku ireverzibilných morfologických zmien parenchýmu v zmysle fibrózy, atrofie acinárnych buniek a kalcifikácie vývodov. Vplyvom uvedených zmien dochádza k poškodeniu exokrinnej aj endokrinnej funkcie pankreasu [2].
Zatiaľ čo u dospelých sa ako hlavný etiologický faktor uvádza nadmerná konzumácia alkoholu, u detí sa okrem anatomických anomálií pankreasu a žlčových ciest uplatňujú predovšetkým genetické faktory. Mutácie v géne PRSS1 (kationický trypsinogén) [3] podmieňujú vznik hereditárnej pankreatitídy (HP), kým mutácie v génoch SPINK1 (inhibítor serínovej proteázy typ Kazal 1) [4], CTRC (chymotrypsín C) [5], CFTR (transmembránový regulátor vodivosti pri cystickej fibróze) [6], CPA1 (karboxypeptidáza A1) [7] a CLDN2 (klaudín 2) [8] sú asociované so zvýšeným rizikom vzniku CHP.
Cieľom tejto štúdie bolo charakterizovať mutačné spektrum génov asociovaných so vznikom CHP u detí s idiopatickou chronickou pankreatitídou (ICHP) a rekurentnou akútnou pankreatitídou (RAP).
Súbor pacientov a metodika
Charakteristika pacientov
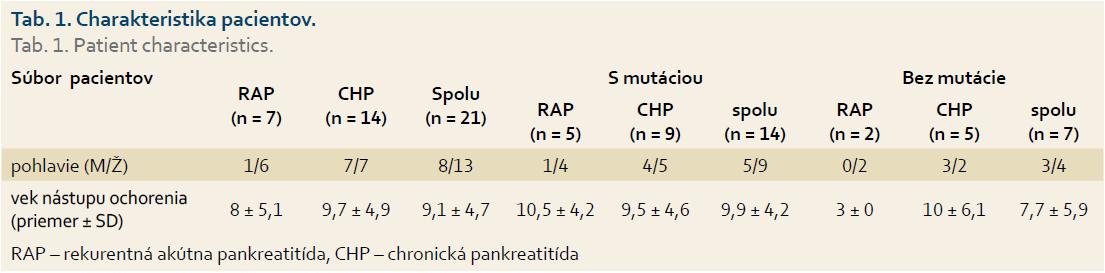
Do štúdie bolo zaradených 21 detských pacientov s RAP alebo ICHP, ktorí boli v období 2008–2013 vyšetrení na II. detskej klinike LF UK a DFNsP Bratislava (tab. 1) alebo ich vzorka krvi bola zaslaná do Laboratória klinickej a molekulovej genetiky II. detskej kliniky LF UK a DFNsP Bratislava za účelom uskutočnenia genetickej analýzy pre podozrenie geneticky podmienenej RAP/ICHP. Diagnóza RAP alebo CHP bola stanovená v súlade s odporúčaním konzorcia INSPPIRE (International Study Group of Pediatric Pancreatitis: In Search for a Cure) [9]. Ako RAP bol označený výskyt minimálne dvoch jednoznačných atakov akútnej pankreatitídy (AP) spolu s: 1. úplným vymiznutím bolesti (≥ jednomesačný bezbolestný interval medzi diagnó-zami AP) alebo 2. úplnou normalizáciou hladín sérových pankreatických enzýmov (amylázy, lipázy) pred nástupom ďalšej epizódy AP. Diagnóza CHP bola stanovená splnením aspoň jedného z uvedených kritérií: 1. abdominálna bolesť a zobrazovacie vyšetrenie potvrdzujúce chronické pankreatické poškodenie; 2. prítomnosť exokrinnej pankreatickej insuficiencie a zobrazovacie vyšetrenie potvrdzujúce chronické pankreatické poškodenie alebo 3. prítomnosť endokrinnej pankreatickej insuficiencie a zobrazovacie vyšetrenie potvrdzujúce chronické pankreatické poškodenie, alebo bioptická vzorka demonštrujúca histopatologické zmeny charakteristické pre CHP. Diagnóza HP bola stanovená v prípade výskytu mutácie v géne PRSS1 u pacienta s pozitívnou rodinnou anamnézou. Jedinci, u ktorých boli vylúčené všetky iné príčiny CHP (trauma, anatomické anomálie pankreasu, lieky, infekcie, systémové a metabolické ochorenia, pozitívna rodinná anamnéza), boli zaradení do skupiny ICHP.
Štúdia bola schválená etickou komisiou DFNsP Bratislava. Rodičia pa- cienta boli informovaní o priebehu štúdie a pacienti boli zaradení na základe rodičmi podpísaného informovaného súhlasu.
Molekulovo-genetická analýza
Periférna krv (2–3 ml) od pacientov bola odobratá do skúmavky EDTA za účelom následnej izolácie genomickej DNA prostredníctvom komerčne dostupného izolačného kitu (Qiagen, Hilden, Germany). Molekulovo-genetická analýza génov PRSS1, SPINK1 a CTRC sa uskutočnila v spolupráci s Oddelením lekárskej genetiky Onkologického ústavu sv. Alžbety. V prípade génov PRSS1 a SPINK1 boli priamym sekvenovaním vyšetrené všetky exóny a exón-intrónové hranice, kým v géne CTRC exóny 2, 3 a 7. V troch prípadoch sa analýza uskutočnila v spolupráci s Ústavem biologie a lékarské genetiky 2. LF UK a FN v Motole, Praha.
Štatistická analýza
Na porovnanie priemerného veku nástupu ochorenia medzi jednotlivými skupinami bol použitý Mann-Whitneyho U test, v ostatných prípadoch bol použitý Fisherov exaktný test. Za štatisticky významný rozdiel bol považovaný p < 0,05.
Výsledky
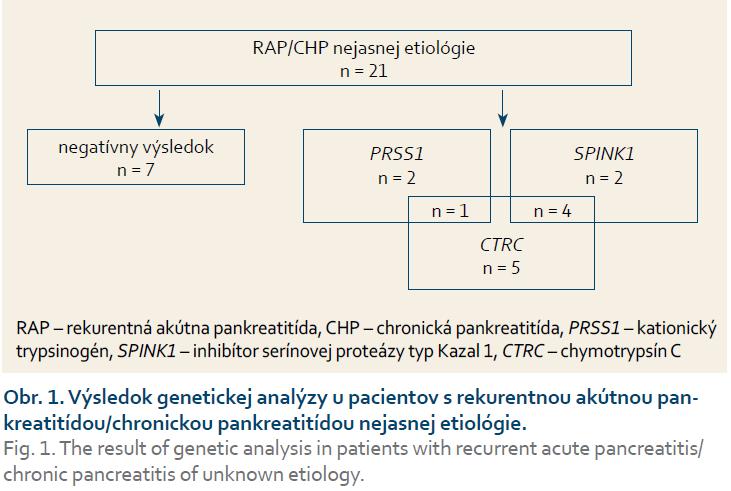
Z celkového počtu 21 detí s diagnózou RAP/CHP u 14 (67 %) bola potvrdená mutácia aspoň v jednom z vyšetrených génov (PRSS1, SPINK1, CTRC) (obr. 1). V troch (14 %) prípadoch bola zistená mutácia v géne PRSS1, u šiestich (28 %) pacientov sa jednalo o mutáciu v géne SPINK1 a u 10 (47 %) o mutáciu v géne CTRC; pričom u piatich pacientov bola súčasne dokázaná mutácia dvoch génov.
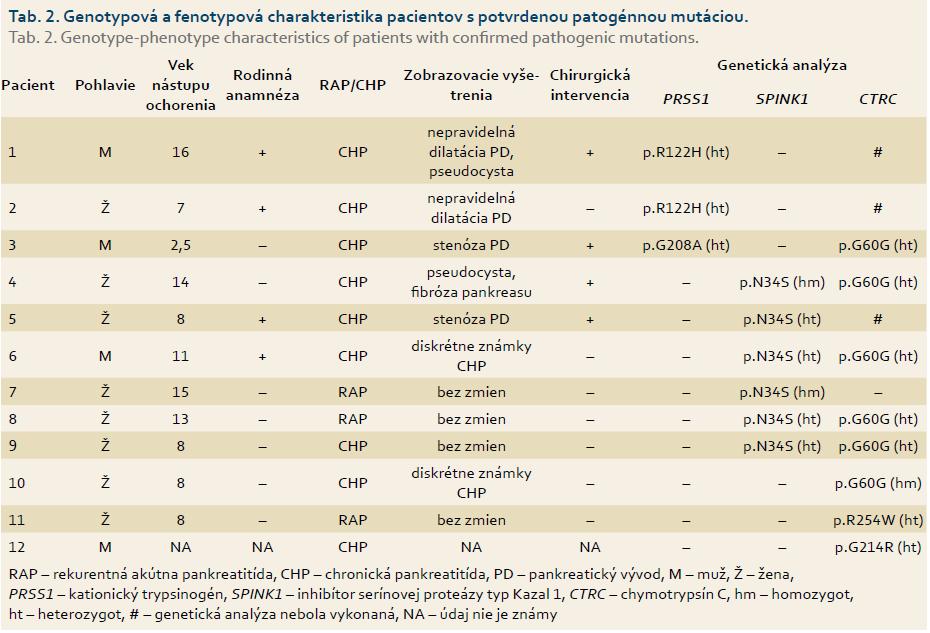
Priemerný vek nástupu ochorenia bol 9,1 ± 4,7 (rozsah od 2,5 do 17), ktorý sa štatisticky významne nelíšil medzi skupinami pacientov s potvrdenou mutáciou a bez nej (9,9 ± 4,2 resp. 7,7 ± 5,92; p = 0,34), ani medzi skupinou chlapcov a dievčat (10,5 ± 6,3 resp. 8,5 ± 4,3; p = 0,51) (tab. 1) a skupinou s diagnózou RAP v porovnaní s CP (8 ± 5,1 resp. 9,7 ± 4,9 (p = 0,51). Diagnóza CHP bola stanovená v 14 (67 %) prípadoch, u siedmich (33 %) detí sa jednalo o RAP. Rodinná anamnéza bola pozitívna v piatich (24 %) prípadoch, z toho v štyroch sa potvrdila genetická príčina ochorenia: v dvoch rodinách bola prítomná mutácia v géne PRSS1 a v dvoch rodinách v géne SPINK1. Chirurgická intervencia bola nevyhnutná celkovo v štyroch prípadoch, pričom u každého dieťaťa bola dokázaná mutácia aspoň v jednom z vyššie uvedených génov (p = 0,26) (tab. 2).
Mutácie v géne PRSS1
Mutácie v géne SPINK1
Mutácie v géne CTRC
Diskusia
Molekulovo-genetická analýza génov PRSS1, SPINK1 a CTRC bola uskutočnená u 21 detských pacientov s diagnózou CHP alebo RAP s cieľom zistiť mutačné spektrum ochorenia v slovenskej populácii.
HP je zriedkavou príčinou CHP. Pri prevalencii 0,3/100 000, ktorá sa udáva v západnej Európe [13], môžeme na Slovensku očakávať približne 17 prípadov HP. Ochorenie je spôsobené mutáciou génu PRSS1, ktorý kóduje kationický trypsinogén. Mutácie typu „gain-of-function“ (mutácie, ktoré vedú k zmene génového produktu v zmysle ich zvýšenej alebo abnormálnej funkcie) génu PRSS1 (p.R122H, p.N29I, p.A16V) sú zodpovedné za veľkú väčšinu prípadov HP kaukazskej populácie. Biochemické štúdie preukázali, že tieto mutácie vedú k zvýšenej auto- aktivácii trypsinogénu a/alebo zvýšenej stabilite trypsínu [14]. Tento nález potvrdzuje hypotézu, podľa ktorej je ochorenie pankreatitída vznikajúce na podklade samonatrávenia žľazy, kedy predčasne aktivovaný trypsín v pankrease hrá kľúčovú úlohu v aktivácii kaskády všetkých pankreatických digestívnych enzýmov [15].
V géne PRSS1 bola prítomná mutácia u troch (14 %) pacientov s RAP/CHP. Frekvencia výskytu mutácie je porovnateľná s nemeckou štúdiou, v ktorej bolo vyšetrených 96 detí s CHP a mutácia v géne PRSS1 sa zistila u 11 (11 %) [4]. Vo švajčiarskej štúdii 16 pacientov so skorým nástupom ochorenia sa PRSS1 mutácia potvrdila u dvoch (12,5 %) [16]. Vyšší výskyt mutácií PRSS1 (23,1 %) u detí s CHP bol popísaný v poľskej štúdii [17]. Mutácia p.R122H bola potvrdená u dvoch pacientov s pozitívnou rodinnou anamnézou. Ide o najčastejšiu mutáciu asociovanú so vznikom HP, ktorá vedie k zámene arginínu za histidín v rámci kodónu 122. Arginín 122 je iniciačným miestom autolytickej autoinaktivácie trypsínu, a preto zámena aminokyselín na tejto pozícii zablokuje účinnú intrapankreatickú inaktiváciu trypsínu [3]. Prvé príznaky ochorenia sa väčšinou objavujú okolo 10. roku života [18], čo môžeme potvrdiť aj v prípade našich pacientov s priemerným vekom nástupu 11,5 rokov. Dedičnosť je autozómovo dominantná s inkompletnou penetranciou (80 %). Rodokmeň pacienta na obr. 2 sa vyznačuje inkompletnou penetranciou, treba si však uvedomiť, že príznaky ochorenia u zdravej nosičky sa vzhľadom na jej vek (12 rokov) ešte môžu prejaviť. U jej súrodencov sa však ochorenie v tomto veku už manifestovalo. U druhého pacienta s mutáciou p.R122H sa ochorenie prejavilo po nezávažnom úraze brucha (úder loptou do brucha pri futbale). Stav bol prvotne uzavretý ako posttraumatická pankreatitída s vývojom pseudocysty a následnými chronickými zmenami na pankrease. Päť mesiacov po prepustení sa však u pacienta vytvoril pankreatický ascites, ktorý vyžadoval chirurgické riešenie. Vzhľadom na pozitívnu rodinnú anamnézu bolo u pacienta doplnené genetické vyšetrenie a potvrdená diagnóza HP [19].
Mutácie génu PRSS1 sa však našli nielen u pacientov s HP, ale aj u pacientov s ICHP [20]. V našom súbore sa v jednom prípade potvrdila mutácia p.G208A v heterozygotnom stave. Uvedená mutácia je veľmi zriedkavá – počet publikovaných prípadov 29 (www.pancreasgenetics.org) – a doteraz bola považovaná za unikátnu mutáciu typickú pre ázijskú populáciu [21]. Mechanizmus, ako uvedená mutácia vedie k vzniku ochorenia, sa odlišuje od mutácie p.R122H. Mutáciou indukovaná zmena proteínu spôsobí jeho hromadenie v bunke, čo vedie k vzniku tzv. stresu endoplazmatického retikula, ktorý v konečnom dôsledku spôsobí poškodenie acinárnych buniek [22]. U pacienta sa okrem tejto mutácie potvrdila prítomnosť mutácie p.G60G v heterozygotnom stave. Rovnaký genotyp p.G208A (PRSS1) /p.G60G (CTRC) mala klinicky zdravá matka a stará matka. Jedným možným vysvetlením pre tento stav by mohla byť nižšia penetrancia mutácie p.G208A alebo prítomnosť ďalších doposiaľ neznámych environmentálnych faktorov, ktoré sa u matky a starej matky pacienta neuplatňujú alebo len mierne [23].
SPINK1 je potencionálny inhibítor proteáz a považuje sa za špecifický inaktivačný faktor intrapankreatickej trypsínovej aktivity [4]. Tento peptid je schopný inaktivovať až 20 % trypsínovej aktivity, a predstavuje tak prvý stupeň ochrany pankreasu pred samonatrávením. Mutácie v géne SPINK1 sa vyskytujú u 1–3 % populácie a pre ich nízku penetranciu sa považujú za predispozičný alebo chorobu modifikujúci a nie za chorobu vyvolávajúci faktor. Najčastejšou mutáciou v géne SPINK1 je p.N34S, ktorého biochemický efekt dodnes nebol objasnený. Asociácia mutácie so vznikom chronickej pankreatitídy však bola potvrdená viacerými štúdiami [4,24].
V našom súbore bola prítomná mutácia p.N34S génu SPINK1 v šiestich (28 %) prípadoch. V súbore amerických pediatrických pacientov bola daná mutácia pozorovaná až v 40 % prípadov [24], podobne vysoký výskyt (43 %) bol popísaný v nemeckej populácii [25]. Nižší výskyt v našej populácii je možné vysvetliť rozdielnou geografickou distribúciou mutácie. Fenotyp pacientov s mutáciou p.N34S bol značne odlišný (tab. 2), pričom v dvoch prípadoch bol pozorovaný rodinný výskyt ochorenia.
Enzým CTRC podporuje proteolytickú inaktiváciu trypsinogénu a trypsínu v podžalúdkovej žľaze, čím predstavuje ďalší dôležitý obranný mechanizmus pred vznikom pankreatitídy. Patogénne mutácie génu CTRC vedú k strate funkcie proteínu odlišnými, avšak vzájomne sa nevylučujúcimi mechanizmami, ktoré znižujú sekréciu, katalytickú aktivitu a proteolytickú stabilitu enzýmu [26].
Mutácia v géne CTRC bola potvrdená u 10 (47 %) pacientov. Najčastejší bol výskyt mutácie p.G60G, ktorá sa zistila u ôsmich pacientov. Jedná sa o synonymný variant (nedochádza k zámene aminokyselín v dôsledku mutácie), ktorý v heterozygotnom stave len veľmi mierne, 1,65× zvyšuje riziko vzniku CHP; v homozygotnej forme až 8,44× [27]. Homozygotná forma mutácie p.G60G bola potvrdená u jednej pacientky s CHP, u ktorej predpokladáme, že vznik ochorenia je asociovaný s danou mutáciou. V dvoch prípadoch sa okrem variantu p.G60G v heterozygotnej forme nezistila ďalšia mutácia v žiadnom z vyšetrovaných génov. Nakoľko je miera zvýšenia rizika vzniku CHP v tejto forme veľmi nízka, u týchto pacientov predpokladáme pôsobenie iného etiologického faktora ako hlavnú príčinu vzniku ochorenia. CHP sa v súčasnosti považuje za komplexné genetické ochorenie, ktoré vzniká interakciou environmentálnych a genetických faktorov [28]. U pacientov s dokázanou mutáciou v génoch PRSS1/SPINK1 a pri súčasnom výskyte mutácie p.G60G v heterozygotnom stave môže preto dôjsť k vzájomnému potencovaniu rizika vzniku ochorenia.
V jednom prípade bola potvrdená mutácia p.R254W v heterozygotnom stave. Mutácia sa vyskytuje približne u 2 % pacientov s CHP [5]. Ide o patogénny variant, u ktorého mutácia génu spôsobí sekrečný defekt proteínu a zároveň dochádza k zvýšenej degradácii proteínu trypsínom [10]. Mutácia p.G214R bola zistená u jedného pacienta v heterozygotnom stave. Ide o unikátny variant, ktorý nebol doposiaľ popísaný v literatúre, jeho klinický efekt bol neznámy. V spolupráci s Ústavom molekulárnej a bunkovej biológie Univerzity v Bostone, USA, bola vykonaná funkčná analýza, ktorá potvrdila patogénnosť mutácie. Na biochemickej úrovni dochádza k zníženiu katalytickej aktivity enzýmu [29].
Limitáciou uvedenej štúdie je relatívne malý súbor pacientov. Táto skutočnosť mohla zohrávať úlohu pri snahe o vytvorenie genotypovo-fenotypovej korelácie. Nepozorovali sme štatisticky významný rozdiel pri porovnaní fenotypov pacientov s mutáciou niektorého génu a bez mutácie.
U pacientov, u ktorých nebola dokázaná prítomnosť mutácie ani v jednom z vyšetrených génov, sa môžu uplatniť iné, zriedkavejšie príčiny ochorenia ako mutácie v géne CPA1 alebo CLDN2, ktoré sú taktiež asociované so vznikom CHP, pričom práve mutácie génu CPA1 boli popísané v súbore pacientov so včasným nástupom ochorenia [7].
Záver
Mutácie génov PRSS1 a SPINK1 sa na Slovensku vyšetrujú na Oddelení lekárskej genetiky Onkologického ústavu sv. Alžbety, Bratislava. V prípade záujmu o vyšetrenie mutácií v géne CTRC je možné vzorku zaslať do Laboratória klinickej a molekulovej genetiky II. detskej kliniky LF UK DFNsP Bratislava.
Autori ďakujú MUDr. Eve Bačinskej (Žilina), MUDr. Renáte Szépeovej, PhD. (Martin), MUDr. Jane Kosnáčovej (Bratislava), MUDr. Jaroslavovi Bibzovi, PhD. (Bratislava) a doc. MUDr. Denise Ilenčíkovej, PhD. (Bratislava) za spoluprácu.
Štúdia bola podporená Grantmi Univerzity Komenského 244/2012 a 573/2013, a grantom Collegium Talentum (EH).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Doručeno/Submitted: 5. 11. 2015
Přijato/Accepted: 29. 11. 2015
MUDr. Ester Hegyi, PhD.
Department of Molecular and Cell Biology
Boston University Medical Campus
72 East Concord Street, Evans-433
Boston, MA 021 18
hester@bu.edu

Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
2. Whitcomb DC. Mechanisms of disease: advances in understanding the mechanisms leading to chronic pancreatitis. Nat Clin Pract Gastroenterol Hepatol 2004; 1 (1): 46–52.
3. Whitcomb DC, Gorry MC, Preston RA et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996; 14 (2): 141–145.
4. Witt H, Luck W, Hennies HC et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000; 25: 213–216.
5. Rosendahl J, Witt H, Szmola R et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 2008; 40 (1): 78–82.
6. Sharer N, Schwarz M, Malone G et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Eng J Med 1998; 339 (10): 645–652.
7. Witt H, Beer S, Rosendahl J et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet 2013; 45 (10): 1216–1220. doi: 10.1038/ng. 2730.
8. Whitcomb DC, LaRusch J, Krasinskas AM et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet 2012; 44 (12): 1349–1354. doi: 10.1038/ng.2466.
9. Morinville VD, Husain SZ, Bai H et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J Pediatr Gastroenterol Nutr 2012; 55 (3): 261–265.
10. Beer S, Zhou J, Szabó A et al. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013; 62 (11): 1616–1624. doi: 10.1136/gutjnl-2012-303090.
11. Comfort MW, Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 1952; 21 (1): 54–63.
12. Čierna I. Hereditárna pankreatitída. Gastroenterol Prax 2009; 8 (2): 94–98.
13. Rebours V, Levy P, Ruszniewski P. An overview of hereditary pancreatitis. Dig Liver Dis 2012; 44 (1): 8–15. doi: 10.1016/j.dld.2011.08.003.
14. Sahin-Tóth M, Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 2000; 278 (2): 286–289.
15. Chiari H. Über die Selbstverdauung des menschlichen Pancreas. Z Heilk 1896; 17: 69–96.
16. Truninger K, Kock J, Wirth HP et al. Trypsinogen gene mutations in patients with chronic or recurrent acute pancreatitis. Pancreas 2001; 22 (1): 18–23.
17. Sobczyńska-Tomaszewska A, Bak D, Oralewska B et al. Analysis of CFTR, SPINK1, PRSS1 and AAT mutations in children with acute or chronic pancreatitis. J Pediatr Gastroenterol Nutr 2006; 43 (3): 299–306.
18. Howes N, Greenhalf W, Rutherford S et al. A new polymorphism for the R122H mutation in hereditary pancreatitis. Gut 2001; 48 (2): 247–250.
19. Čierna I, Cingel V. Hereditárna pankreatitída u 17-ročného pacienta. Pediatr Prax 2011; 12 (3): 121–122.
20. Bátovský M. Úloha génových mutácií v patogenéze chronickej pankreatitídy. Gastroenterol Prax 2013; 12 (2): 72–74.
21. Masamune A, Nakano E, Kume K et al. PRSS1 c.623G>C (p.G208A) variant is associated with pancreatitis in Japan. Gut 2014; 63 (2): 366. doi: 10.1136/gutjnl-2013- 304925.
22. Schnúr A, Beer S, Witt H et al. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut 2014; 63 (2): 337–343. doi: 10.1136/gutjnl-2012- 304331.
23. Hegyi E, Čierna I, Vavrová Ľ et al. Chronic pancreatitis associated with the p.G208A variant of PRSS1 gene in a European patient. JOP 2014; 15 (1): 49–52. doi: 10.6092/1590-8577/1896.
24. Pfützer RH, Barmada MM, Brunskill AP et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000; 119 (3): 615–623.
25. Truninger K, Witt H, Köck J et al. Mutations of the serine protease inhibitor, Kazal type 1 gene, in patients with idiopathic chronic pancreatitis. Am J Gastroenterol 2002; 97 (5): 1133–1137.
26. Zhou J, Sahin-Tóth M. Chymotrypsin C mutations in chronic pancreatitis. J Gastroenterol Hepatol 2011; 26 (8): 1238–1246. doi: 10.1111/j.1440-1746.2011.06791.x.
27. Beer S, Ludwig M, Németh BC et al. Synonymous CTRC variant c.180C>T contributes significantly to chronic pancreatitis risk. Pancreatology 2014; 14 (3): S15.
28. Rosendahl J, Landt O, Bernadova J et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut 2013; 62 (4): 582–592. doi: 10.1136/gutjnl-2011-300 645.
29. Szabó A, Ludwig M, Hegyi E et al. Mesotrypsin signature mutation in a chymotrypsin C (CTRC) variant associated with chronic pancreatitis. J Biol Chem 2015; 290 (28): 17282–17292. doi: 10.1074/jbc.M114.618439.