Smíšený adenoneuroendokrinní karcinom žaludku – kazuistika
Marek Drab1, Eva Tomsová1, Juraj Marček1, Tomáš Klinger2
+ Pracoviště
Souhrn
Neuroendokrinní tumory představují heterogenní skupinu neoplazií vycházejících z různých anatomických lokalizací, přičemž přibližně 50 % je gastrointestinálního původu. Mezi klíčové parametry při hodnocení každého případu patří morfologie nádoru, počet mitotických buněk a index Ki-67. Smíšené adenoneuroendokrinní karcinomy (MANEC) jsou vzácné agresivní novotvary sestávající z adenokarcinomatózních i neuroendokrinních buněk, přičemž každá složka musí tvořit alespoň 30 % léze. Naším případem je 77letý polymorbidní pacient, který pro známky akutního krvácení do horní oblasti gastrointestinálního traktu s vyjádřeným anemickými syndromem podstoupil gastroskopické vyšetření s nálezem vředové léze na přední stěně žaludku na přechodu těla a antra. Při kontrolních gastroskopických vyšetřeních s odběrem biopsií byly nejdříve histologicky prokázány pouze známky chronické gastritidy při pozitivitě na Helicobacter pylori, následně nalezeny fragmenty high-grade tubulárního až tubulovilózního adenomu a struktury středně diferencovaného tubulárního adenokarcinomu. Histologickým rozborem žaludečního resekátu byl prokázán smíšený adenoneuroendokrinní karcinom s lymfangioinvazí.
Klíčová slova
neuroendokrinní tumor, smíšený adenoneuroendokrinní karcinom, MANEC, synaptofyzin, chromogranin
Úvod
Neuroendokrinní tumory představují heterogenní skupinu neoplazií vycházejících z různých anatomických lokalizací, přičemž přibližně 50 % je gastrointestinálního původu [1–3]. Incidence gastroenteropankreatických neuroendokrinních tumorů v Evropě se pohybuje v rozmezí 1,3–2,3/100 tis. obyvatel. Data ale pocházejí z jednotlivých národních registrů se značnou heterogenitou [4–6]. Neuroendokrinní tumory se mohou vyvinout v kterékoli části gastrointestinálního traktu, nejčastěji se vyskytují v tenkém střevě (45 %), dále pak v rektu (20 %), appendixu (16 %), tlustém střevu (11 %) a žaludku (7 %) [7]. Většina neuroendokrinních tumorů se klinicky projevuje jako metastatické onemocnění nebo k němu progreduje s průměrným přežitím 3 roky [1]. To představuje kontrast s obecně vnímanou představou neuroendokrinních tumorů jako pomalu progredujících malignit, které často nevyžadují léčbu. Neuroendokrinní tumory se obecně vyznačují indolentními, ale vysoce variabilními klinickými projevy. Mezi klíčové parametry při hodnocení každého případu patří morfologie nádoru, počet mitotických buněk a index Ki-67. Ačkoli většina gastrointestinálních neuroendokrinních tumorů je nesekrečních, u některých pacientů se vyskytnou nebo se u nich rozvinou sekreční syndromy [8]. Vzhledem k inaktivaci serotoninu v játrech se karcinoidový syndrom obvykle vyskytuje při metastatickém postižení jater. Klinicky se projevuje návaly horka, sekrečním průjmem a dušností. Další méně časté sekreční syndromy se vyskytují u gastrinomů průjmem a peptickými ulceracemi, u ghrelinomů anorexií a hubnutím, u VIPomů vodnatým průjmem a sklonem k hypokalemii či acidóze, u somatostatinomů diabetem, průjmem, steatoreou, zvýšeným výskytem cholelitiázy a u neurotenzinomů edémem, sklonem k hypotenzi, cyanóze a výskytem návalů horka. U nesekrečních neuroendokrinních tumorů mohou klinické projevy vzniknout v důsledku lokálního postižení, např. epizodickými bolestmi břicha, projevy obstrukce v důsledku mezenterické fibrózy nebo střevní ischemie, ascitem, anemií či projevy malabsorpce. Projevy jaterní dysfunkce obvykle doprovází metastatické postižení jater [9–11]. Většina gastrointestinálních neuroendokrinních tumorů je zjištěna náhodně v rámci diagnostiky anemie, dyspeptických obtíží, bolestí břicha, hmotnostního úbytku, vzácně krvácení do gastrointestinálního traktu s podílem přibližně 0,18 % všech možných příčin krvácení [12–14].
Diagnostika
Všichni pacienti by v počáteční fázi diagnostiky a v průběhu onemocnění měli být komplexně vyšetřeni s cílem objasnit symptomy potenciálně související se sekrečním syndromem. Laboratorním vyšetřením by se měla potvrdit, nebo vyloučit hypersekrece peptidů. U pacientů se symptomy připomínajícími karcinoidový syndrom by měla být provedena analýza kyseliny 5-hydroxyindoloctové ve 24hodinovém sběru moči. Chronické zvýšení cirkulujícího serotoninu může také vést ke vzniku karcinoidového srdečního onemocnění s chlopenní dysfunkcí pravého srdce a následným projevem srdečního selhání až ke smrti, proto je u těchto pacientů důležité echokardiografické vyšetření [8,10,15]. Nezbytnou součástí diagnostiky neuroendokrinních tumorů je histologická verifikace ideálně metodou aspirace tenkou jehlou pro lepší dostupnost materiálu pro analýzu. Při histologickém podezření na neuroendokrinní tumor je doplněno imunohistochemické vyšetření k průkazu nízkomolekulárních keratinů, chromograninu a synaptofyzinu. Stanovení indexu Ki-67 by mělo být provedeno ve všech okrscích vyšetřovaného materiálu a v oblastech s nejvyšší mitotickou hustotou vzhledem k intratumorální heterogenitě [16]. Dobře diferencované neuroendokrinní karcinomy mají obvykle index Ki-67 v rozmezí 20–55 %, zatímco špatně diferencované velkobuněčné nebo malobuněčné neuroendokrinní karcinomy mají obvykle index Ki-67 > 55 % [17]. V případě neznámé lokality primárního neuroendokrinního tumoru nebo u keratin negativního nádoru je doporučeno imunohistochemické vyšetření k průkazu dalších transkripčních faktorů: TTF-1 (thyroid transcription factor-1), CDX-2 (caudal type homeobox 2), PDX-1 (pancreatic duodenal homeo- box 1) nebo ISL-1 (Islet-1) a PSAP (prostate-specific acid phosphatase) [18–22]. Pozitivita TTF-1, dále imunohistochemická pozitivita na kalcitonin a karcinoembryonální antigen odliší medulární karcinom štítné žlázy od plicního neuroendokrinního tumoru. CDX-2 pozitivita a pozitivní barvení na serotonin představují střevní enterochromafinní endokrinní buňky. Pozitivita ISL-1/PDX-1 a imunohistochemický průkaz pankreatických hormonů poukazuje na pankreatický původ neuroendokrinního tumoru. Pozitivní barvení PSAP a imunohistochemická pozitivita pro GLP-1 (glucagon-like peptide-1) /PP (pancreatic polypeptide) /PYY (peptide YY) se vyskytují u rektálního neuroendokrinního tumoru [23].
Paragangliom je prokázán pozitivitou tyrosin hydroxylázy při keratin negativitě [24]. V rámci diagnostického procesu a průběžného sledování efektivity terapie představují důležitou roli zobrazovací metody. Při pátrání po primárním tumorózním ložisku mají zásadní postavení endoskopie, endoskopická ultrasonografie, CT vyšetření, magnetická rezonance, event. enterografie/enteroklýza [25]. Schopnost neuroendokrinních tumorů vychytávat a dekarboxylovat aminové prekurzory lze využít pro zobrazení těchto tumorů pomocí pozitronové emisní tomografie (PET). PET využívající katecholaminový prekurzor 18F-DOPA (18Fluoro-L-dihydroxyfenylalanin) představuje vysoce citlivou zobrazovací metodu pro neuroendokrinní gastrointestinální nádory [26–28]. 18F-DOPA je transportován do buňky pomocí přenašeče LAT (L-type amino acid transporter) a poté dekarboxylován pomocí AADC (aromatic L-amino acid decarboxylase) za vzniku 18F-dopaminu, který je ná- sledně uložen do specifických zásobních vezikul a chráněn před enzymatickou degradací [29]. Další diagnostickou zobrazovací metodou je pozitronová emisní tomografie 68Ga značenými somatostatinovými receptory, která prokazuje vysokou senzitivitu (93 %; 95% CI 91–95 %) a specificitu (91 %; 95% CI 82–97 %) pro neuroendokrinní tumory s omezenou dostupností [30]. Použití 18F-FDG (18fluoro-fluorodeoxyglukóza) PET/CT je vhodné pro grade 2 a grade 3 neuroendokrinní tumory, které mají vyšší metabolizmus glukózy a menší expresi somatostatinových receptorů ve srovnání s neuroendokrinními tumory nižšího stupně [31]. Somatostatinová receptorová scintigrafie by měla být v rámci diagnostiky použita pouze v případě nedostupnosti PET/CT vzhledem k nižší citlivosti. Pro hodnocení jaterního postižení je preferována MR s aplikací kontrastní látky před vícefázovou CT vzhledem k vyšší senzitivitě a specificitě, obzvlášť u pacientů, u kterých je zvažována ablační či debulking terapie [32,33].
Terapie
Chirurgická, lokoregionální a farmakologická terapie představují základní léčebné modality pro pacienty s neuroendokrinními tumory. Pro optimalizaci léčebných výsledků je důležitá individualizace léčby pacienta multidisciplinárním týmem specialistů. Před definitivním rozhodnutím o léčbě je zapotřebí přehodnotit rozsah a lokalizaci onemocnění, stupeň nádorového postižení, tempo progrese onemocnění, výkonnostní stav, komorbidity a samozřejmě preference pacienta. U primárních žaludečních neuroendokrinních tumorů by měl být při sestavování chirurgického plánu zvažován podtyp onemocnění definovaný klinickými a patologickými rysy [34]. Preferencí jsou minimálně invazivní chirurgické techniky zachovávající objem a funkci žaludku [35]. V zásadě by se sekreční a nesekreční neuroendokrinní tumory měly léčit podobně, se snahou o individualizaci léčebných plánů [36,37]. Pacienti s generalizovaným nebo neresekabilním gastrointestinálním neuroendokrinním tumorem bez známek karcinoidového syndromu by v počáteční fázi terapie měli být léčeni somatostatinovými analogy nebo cílenou (biologickou) léčbou [38,39]. U pacientů s progresí onemocnění na terapii somatostatinovými analogy by měla být zvážena změna terapie na everolimus v monoterapii nebo v kombinaci se somatostatinovými analogy. V současné době neexistuje optimální léčebná strategie léčby 2. linie při selhání terapie 1. linie [38–41]. Fáze III studie PROMID srovnávající somatostatinová analoga (oktreotid LAR) s placebem u naivních pacientů potvrdila antiproliferační aktivitu somatostatinových analog. Doba do progrese tumoru představovala 8,3 měsíců (14,3 vs. 6,0 měsíce; HR = 0,34, 95% CI 0,20–0,59; p = 0,000072) [38,39,42]. Ve studii RADIANT-4 srovnávající everolimus s placebem u pacientů s nesekreční plicní nebo gastrointestinální formou neuroendokrinního tumoru po předchozí terapii somatostatinovými analogy nebo chemoterapii bylo prokázáno prodloužení doby nemoci bez progrese ve prospěch everolimu o 7,1 měsíce (11,0 vs. 3,9 měsíce; HR = 0,48; 95% CI 0,35–0,67; p < 0,00001) [43]. Peptid-receptor radionuklidová terapie (PRRT) se používá více než dvě desetiletí při léčbě neuroendokrinních tumorů. Nová generace PRRT využívá 90Y nebo 177Lu značené vysoce afinitní somatostatinová analoga (oktreotid nebo oktreotát) a stabilnější chelatační látky [44]. Peptid-receptor radionuklidovou terapii s 177Lu lze zvažovat u pacientů s progredujícím, dobře diferencovaným neuroendokrinním tumorem gastrointestinálního traktu (primárně při postižení střeva) s pozitivitou na somatostatinové receptory, Ki-67 < 20 %, bez ohledu na sekreční aktivitu, nereagujícím na terapii somatostatinovými analogy [45].
Smíšené adenoneuroendokrinní karcinomy
Smíšené adenoneuroendokrinní karcinomy (MANEC) jsou vzácné agresivní novotvary s velmi špatnou prognózou. Přesná data týkající se výskytu pacientů s MANEC nejsou známa, dominují kazuistická sdělení. Incidence MANEC v gastrointestinálním traktu se odhaduje na 1–2/1 000 000 obyvatel [46]. Smíšené adenoneuroendokrinní karcinomy představují vzácný podtyp neuroendokrinního novotvaru sestávající z adenokarcinomatózních i neuroendokrinních buněk. Každá složka musí tvořit alespoň 30 % léze [47]. Smíšené adenoneuroendokrinní karcinomy se mohou vyskytovat v různých částech gastrointestinálního traktu. Makroskopicky se tyto novotvary jeví jako polypoidní nebo ulcerující útvary velikosti až několik centimetrů [48]. Často se z endoskopických bioptických vzorků iniciálně diagnostikuje pouze adenokarcinomová složka [49]. Ve srovnání se žaludečními adenokarcinomy mají žaludeční smíšené adenoneuroendokrinní karcinomy horší prognózu a tendenci k metastatickému šíření. Momentálně chybí optimální strategie sledování a terapie u nádorů pronikajících do subserózy, hlubších vrstev a s metastatickým rozsevem [50].
Kazuistika
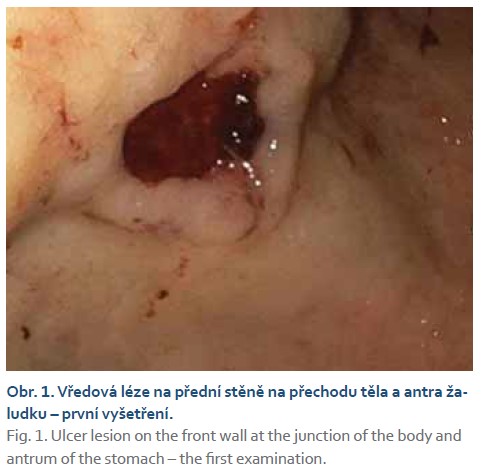
Naším vyšetřovaným je 77letý polymorbidní pacient (anamnesticky ischemická choroba srdeční, stav po infarktu myokardu, permanentní fibrilace síní, arteriální hypertenze, diabetes mellitus druhého typu s chronickými komplikacemi), který byl hospitalizován na jednotce intenzivní péče interního oddělení pro známky akutního krvácení do horní oblasti gastrointestinálního traktu s vyjádřeným anemickými syndromem. Při urgentním gastroskopickém vyšetření byla nalezena hluboká vředová léze na přední stěně žaludku v místě přechodu těla a antra, spodina vředu byla krytá koagulem s navalitými okraji (obr. 1) velikosti přibližně 15 mm (Forrest IIb). Léze byla endoskopicky ošetřena injektorovou aplikací adrenalinu (ředění 1: 10 000) a kontaktní termickou koagulací. Již před endoskopickým vyšetřením byla u pacienta zahájena terapie inhibitory protonové pumpy (omeprazol) v dávce 80 mg bolusově a následována kontinuální intravenózní aplikací v dávce 8 mg/hod po dobu 72 hod, následně v dávce 40 mg denně.
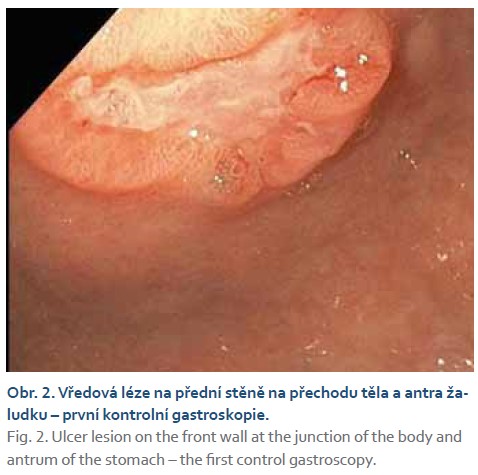
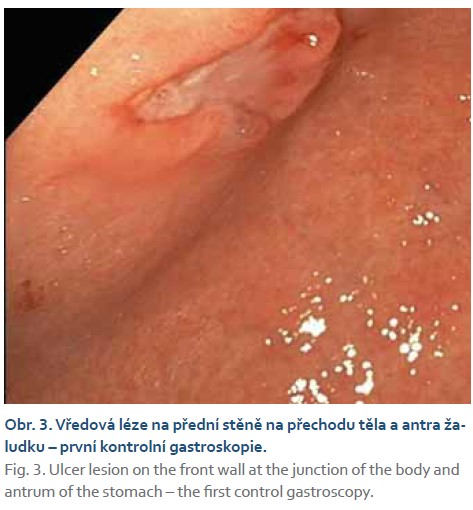
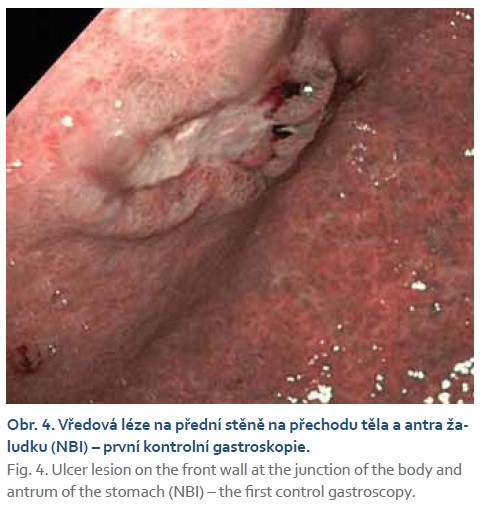
Při kontrolním gastroskopickém vyšetření za 6 týdnů od vstupního vyšetření byla nalezena vředová léze stejné velikosti (obr. 2–4) se spodinou krytou fibrinem (Forrest III). Odběrem biopsie z okraje léze byli prokázány pouze známky chronické gastritidy a přítomnost Helicobacter pylori s následnou eradikací antibiotiky dle standardu.
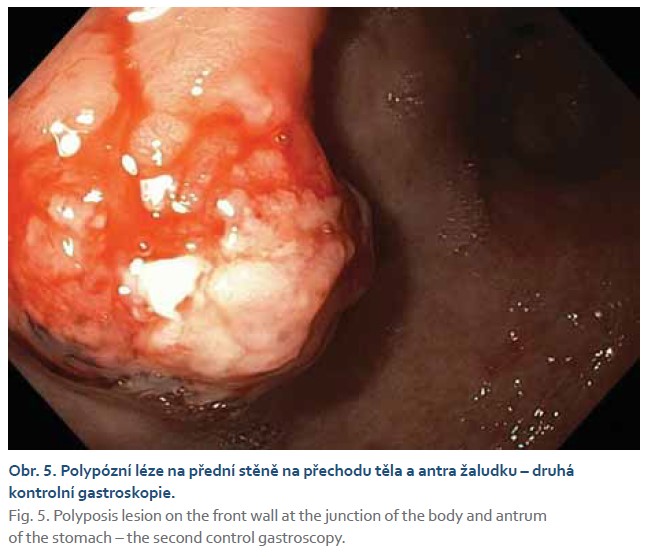
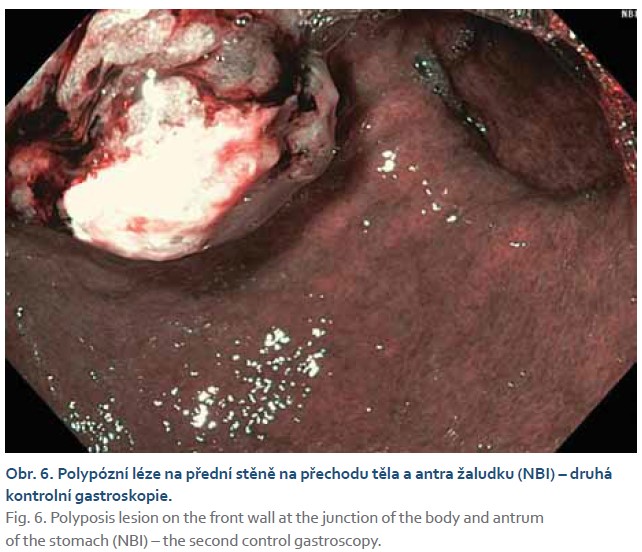
Při další gastroskopické kontrole za 6 týdnů byla nalezena žaludeční polypózní léze stejné lokalizace a velikosti, kontaktně krvácející, na povrchu s přítomností hlenu (obr. 5, 6). Byla provedena další biopsie.
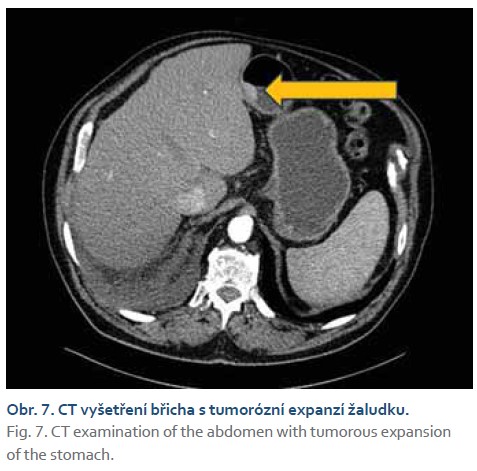
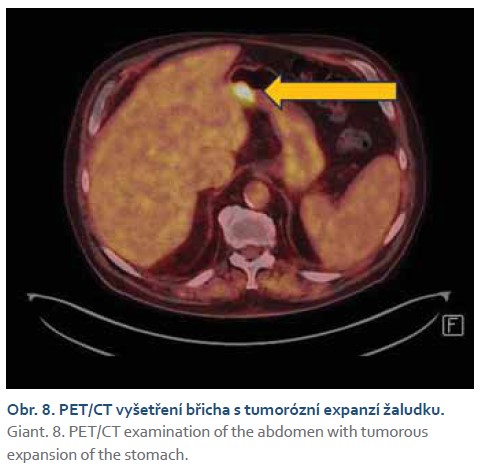
Histologicky se prokázaly fragmenty high-grade tubulárního až tubulovilózního adenomu a v jednom fragmentu i struktury středně diferencovaného tubulárního adenokarcinomu. V rámci stagingu bylo doplněno CT vyšetření s nálezem tumorózní expanze žaludku, hodnoceno T3N0Mx (obr. 7). Pro nález nespecifických plicních nodulů na CT vyšetření bylo doplněno PET/CT, které potvrdilo pouze nález v oblasti žaludku z předchozího CT vyšetření, bez průkazu metastatických plicních ložisek (obr. 8).
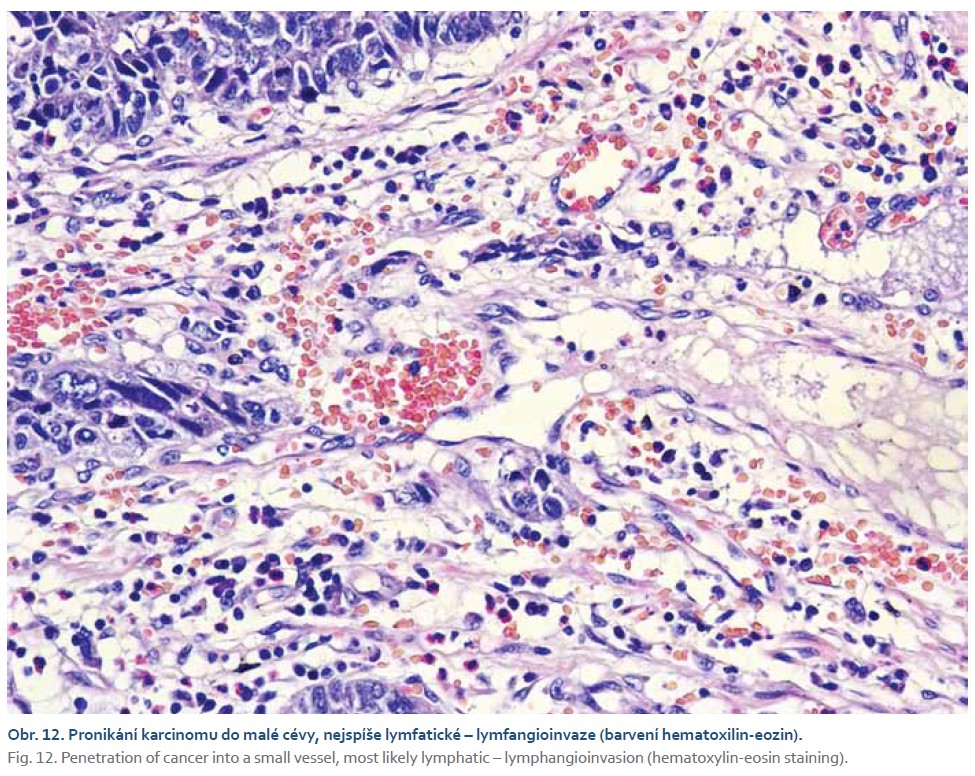
Pacient byl odeslán na vyšší pracoviště, kde podstoupil subtotální gastrektomii s Y-Roux rekonstrukcí. V žaludečním resekátu byl histologicky prokázán smíšený adenoneuroendokrinní karcinom s lymfangioinvazí a imunohistochemickým průkazem neuroendokrinního markeru synaptofyzinu (obr. 9–12). Dominantně převažovala adenokarcinomová složka. Pacient podstoupil adjuvantní chemoterapie režimem fluorouracil, leucovorin, oxaliplatina (FLO). Dosavadní kontrolní CT vyšetření a gastroskopie byly bez známek recidivy tumoru.
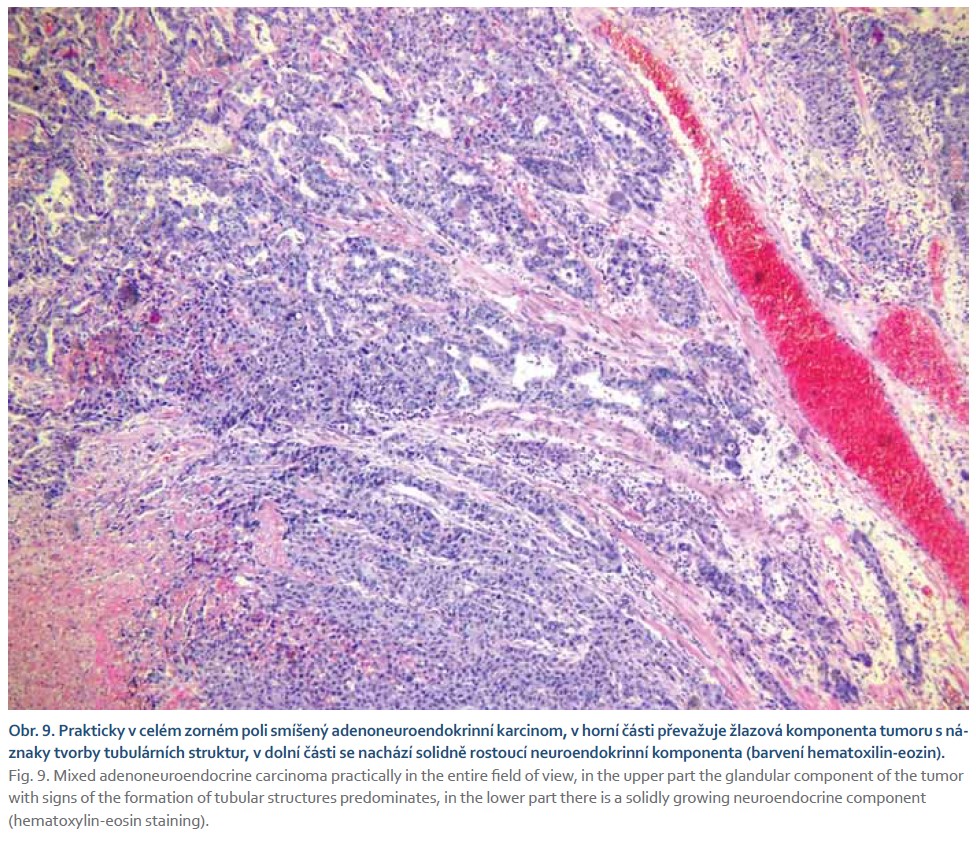
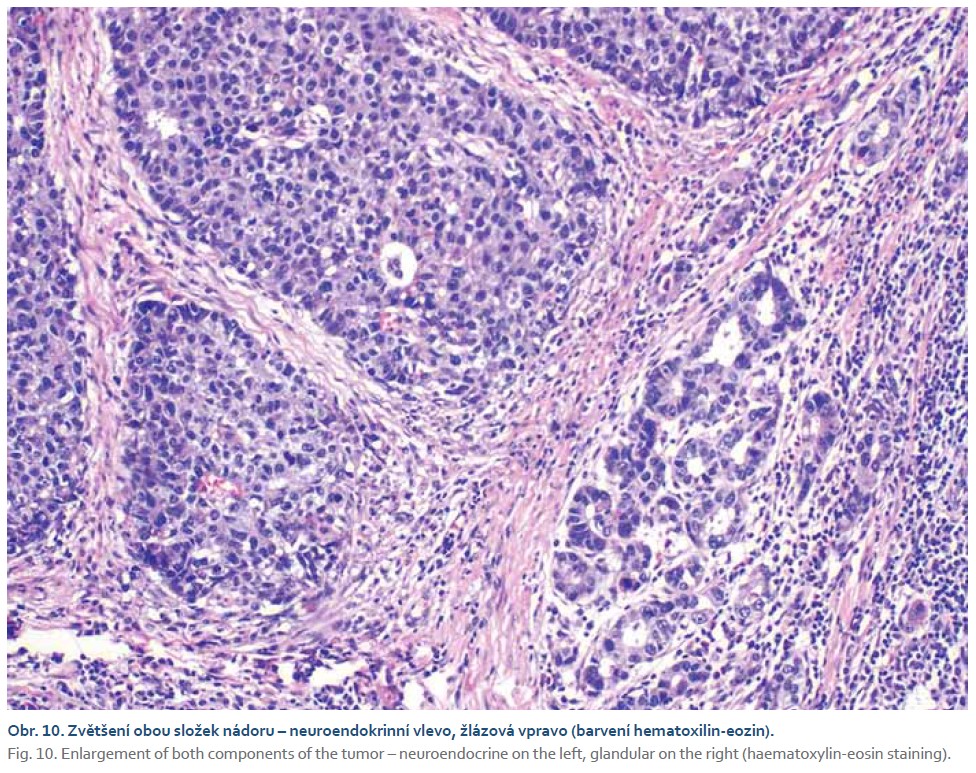
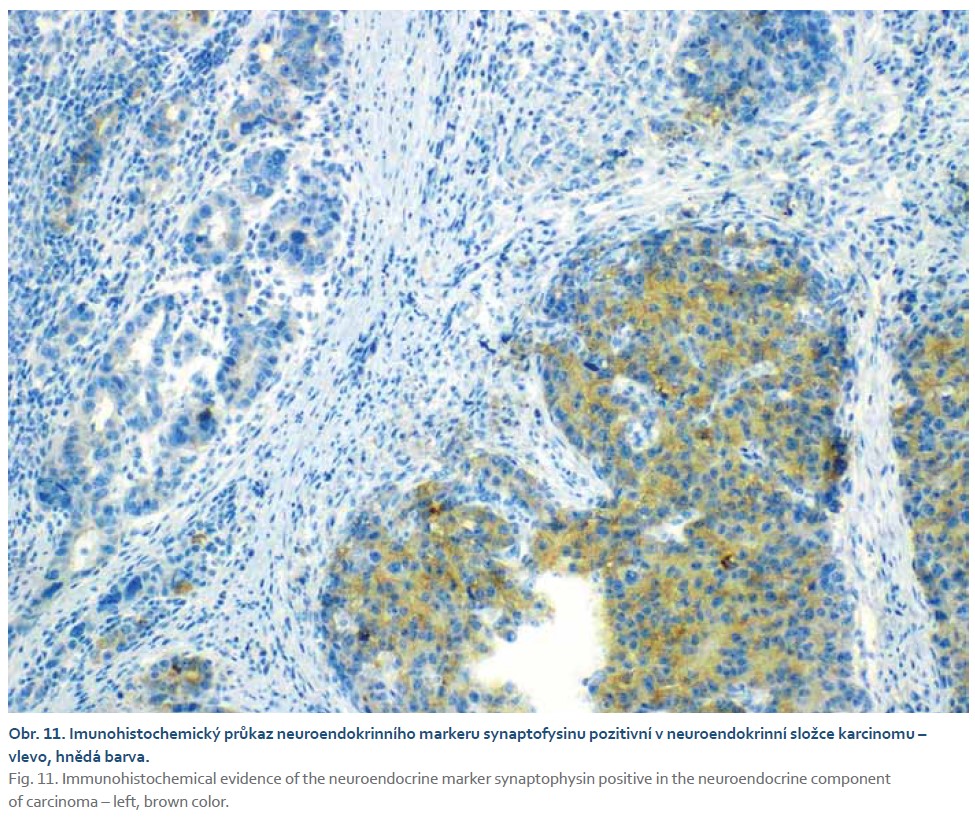
Diagnostika smíšených adenoneuroendokrinních karcinomů se opírá především o histopatologické vyšetření. V případě přítomnosti neuroendokrinní i adenokarcinomatózní složky při barvení hematoxylinem a eozinem je třeba myslet i na možnost smíšeného adenoneuroendokrinního karcinomu. Dalším krokem pro potvrzení diagnózy je imunohistochemické vyšetření s pozitivitou na synaptofyzin, chromogranin nebo CD56. K potvrzení diagnózy je zapotřebí přítomnost alespoň dvou ze tří markerů. Histologicky je neuroendokrinní složka podobná jak malobuněčnému, tak velkobuněčnému neuroendokrinnímu karcinomu plic. Malobuněčná složka je tvořena malými buňkami s řídkou cytoplazmou a fusiformními jádry. Velkobuněčná složka je tvořena buňkami s hojnou cytoplazmou a vezikulárními jádry. Ne-neuroendokrinní část smíšeného adenoneuroendokrinního karcinomu může být tvořena tubulovilózním nebo vilózním adenomem, adenokarcinomem, vzácně spinocelulárním karcinomem [47,51–53]. Molekulární rysy tohoto onemocnění stále nejsou dostatečně prozkoumány. Téměř všechny případy vykazují alespoň jednu somatickou mutaci nad 5 % tkáně. Nejčastěji prokázané mutované geny představují KRAS a TP53 v obou složkách (neuroendokrinní i ne-neuroendokrinní). Mutace genů PI3K a RB1 byla prokázaná pouze v neuroendokrinních složce [54]. Dle studie zahrnující 80 pacientů bylo zjištěno, že celkové 3leté přežití u pacientů s dominantní adenokarcinomovou složkou bylo vyšší a zároveň byla prokázána nižší míra recidivy ve srovnání s neuroendokrinně dominantním tumorem (75 vs. 40 %; p = 0,006). Bylo také zjištěno, že neuroendokrinně dominantní nádory jsou nezávislým rizikovým faktorem pro celkově horší přežití [55]. Obecně je chirurgická resekce indikována u pacientů bez metastatického postižení a může být kurativní u < 30 % pacientů. Vzhledem k vysoké míře recidivy po operaci je adjuvantní chemoterapie léčbou první volby s mediánem přežití v rozmezí od 6 do 12 měsíců. Momentálně chybí jednotná léčebná strategie pro gastrointestinální smíšené adenoneuroendokrinní karcinomy. Omezené informace dostupné pouze z kazuistik a malých retrospektivních studií naznačují, že hlavními terapeutickými strategiemi jsou operace nebo nejednotná chemoradioterapie [55–59].
Závěr
Smíšené adenoneuroendokrinní karcinomy představují agresivní novotvary sestávající z adenokarcinomatózních i neuroendokrinních buněk, přičemž každá složka musí tvořit alespoň 30 % léze. Neuroendokrinně dominantní nádory mají horší prognózu. I přes snahu o kurativní chirurgickou resekci lokálních nálezů je míra recidivy vysoká. Přestože chybí jednotné doporučení pro terapii smíšených adenoneuroendokrinních karcinomů, multidisciplinární přístup k těmto pacientům vede ke snaze o optimalizaci diagnostiky, managementu onemocnění a strategie pravidelných kontrol ke zhodnocení přínosu terapie.
Doručeno/Submitted: 22. 7. 2023
Přijato/Accepted: 31. 8. 2023
MUDr. Marek Drab
Gastroenterologické oddělení
Oblastní nemocnice Mladá Boleslav, a. s.,
nemocnice Středočeského kraje
třída Václava Klementa 147
293 01 Mladá Boleslav
marek.drab@onmb.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Hallet J, Law CH, Cukier M et al. Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer 2015; 121 (4): 589–597. doi: 10.1002/cncr.29099.
2. Oberg K, Castellano D. Current knowledge on diagnosis and staging of neuroendocrine tumors. Cancer Metastasis Rev 2011; 30(1): 3–7. doi: 10.1007/s10555-011-9292-1.
3. Fraenkel M, Kim MK, Faggiano A et al. Epidemiology of gastroenteropancreatic neuroendocrine tumours. Best Pract Res Clin Gastroenterol 2012; 26(6): 691–703. doi: 10.1016/j.bpg.2013.01.006.
4. Fraenkel M, Kim M, Faggiano A et al. Incidence of gastroenteropancreatic neuroendocrine tumours: a systematic review of the literature. Endocr Relat Cancer 2014; 21(3): eR153–eR163. doi: 10.1530/ERC-13-0125.
5. Leoncini E, Boffetta P, Shafir M et al. Increased incidence trend of low-grade and high-grade neuroendocrine neoplasms. Endocrine 2017; 58(2): 368–379. doi: 10.1007/s12020-017-1273-x.
6. Huguet I, Grossman AB, O’Toole D. Changes in the epidemiology of neuroendocrine tumours. Neuroendocrinology 2017; 104(2): 105–111. doi: 10.1159/000441897.
7. Maggard MA, O‘Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240(1): 117–122. doi: 10.1097/01.sla.0000129342.67174.67.
8. Modlin IM, Oberg K, Chung DC et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9(1): 61–72. doi: 10.1016/S1470-2045(07)70410-2.
9. Oberg K, Knigge U, Kwekkeboom D et al. Neuroendocrine gastro-entero- pancreatic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23(7): 124–130. doi: 10.1093/annonc/mds295.
10. Kulke MH, Mayer RJ. Carcinoid tumors. N Engl J Med 1999; 340(11): 858–868. doi: 10.1056/NEJM199903183401107.
11. Schnirer II, Yao JC, Ajani JA. Carcinoid – a comprehensive review. Acta Oncol 2003; 42(7): 672–692. doi: 10.1080/02841860310010547.
12. Sato Y. Endoscopic diagnosis and management of type I neuroendocrine tumors. World J Gastrointest Endosc 2015; 7(4): 346–353. doi: 10.4253/wjge.v7.i4.346.
13. Rindi G, Klöppel G. Endocrine tumors of the gut and pancreas tumor biology and classification. Neuroendocrinology 2004; 80(Suppl 1): 12–15. doi: 10.1159/000080733.
14. Sheibani S, Kim JJ, Chen B et al. Natural history of acute upper GI bleeding due to tumours: short-term success and long-term recurrence with or without endoscopic therapy. Aliment Pharmacol Ther 2013; 38(2): 144–150. doi: 10.1111/apt.12347.
15. Bhattacharyya S, Davar J, Dreyfus G et al. Carcinoid heart disease. Circulation 2007; 116(24): 2860–2865. doi: 10.1161/CIRCULATIONAHA.107.701367.
16. Klimstra DS, Modlin IR, Adsay NV et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol 2010; 34(3): 300–313. doi: 10.1097/PAS.0b013e3181ce1447.
17. Sorbye H, Welin S, Langer SW et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol 2013; 24(1): 152–160. doi: 10.1093/annonc/mds276.
18. Chan ES, Alexander J, Swanson PE et al. PDX-1, CDX-2, TTF-1, and CK7: a reliable immunohistochemical panel for pancreatic neuroendocrine neoplasms. Am J Surg Pathol 2012; 36(5): 737–743. doi: 10.1097/PAS.0b013e31824aba59.
19. Graham RP, Shrestha B, Caron BL et al. Islet-1 is a sensitive but not entirely specific marker for pancreatic neuroendocrine neoplasms and their metastases. Am J Surg Pathol 2013; 37(3): 399–405. doi: 10.1097/PAS.0b013e31826f042c.
20. Srivastava A, Hornick JL. Immunohistochemical staining for CDX-2, PDX-1, NESP-55, and TTF-1 can help distinguish gastrointestinal carcinoid tumors from pancreatic endocrine and pulmonary carcinoid tumors. Am J Surg Pathol 2009; 33(4): 626–632. doi: 10.1097/PAS.0b013e31818d7d8b.
21. Vinik AI, Woltering EA, Warner RR et al. NANETS consensus guidelines for the diagnosis of neuroendocrine tumor. Pancreas 2010; 39(6): 713–734. doi: 10.1097/MPA.0b013e3181ebaffd.
22. Uccella S, Sessa F, La Rosa S. Diagnostic approach to neuroendocrine neoplasms of the gastrointestinal tract and pancreas. Turk Patoloji Derg 2015; 31(Suppl 1): 113–127. doi: 10.5146/tjpath.2015.01319.
23. Kim JY, Kim KS, Kim KJ et al. Non-L-cell immunophenotype and large tumor size in rectal neuroendocrine tumors are associated with aggressive clinical behavior and worse prognosis. Am J Surg Pathol 2015; 39(5): 632–643. doi: 10.1097/PAS.0000000000000400.
24. Tsolakis AV, Grimelius L, Granerus G et al. Histidine decarboxylase and urinary methylimidazoleacetic acid in gastric neuroendocrine cells and tumours. World J Gastroenterol 2015; 21(47): 13240–13249. doi: 10.3748/wjg.v21.i47.13240.
25. Ganeshan D, Bhosale P, Yang T et al. Imaging features of carcinoid tumors of the gastrointestinal tract. AJR Am J Roentgenol 2013; 201(4): 773–786. doi: 10.2214/AJR.12.9758.
26. Koopmans KP, de Vries EG, Kema IP et al. Staging of carcinoid tumours with 18F-DOPA PET: a prospective, diagnostic accuracy study. Lancet Oncol 2006; 7(9): 728–734. doi: 10.1016/S1470-2045(06)70801-4.
27. Koopmans KP, Neels OC, Kema IP et al. Improved staging of patients with carcinoid and islet cell tumors with 18F-dihydroxy-phenyl-alanine and 11C-5-hydroxy-tryptophan positron emission tomography. J Clin Oncol 2008; 26(9): 1489–1495. doi: 10.1200/JCO.2007.15.1126.
28. Hoegerle S, Altehoefer C, Ghanem N et al. Whole-body 18F dopa PET for detection of gastrointestinal carcinoid tumors. Radiology 2001; 220(3): 373–380. doi: 10.1148/radiology. 220.2.r01au25373.
29. Eisenhofer G, Huynh TT, Hiroi M et al. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord 2001; 2(3): 297–311. doi: 10.1023/A: 1011572617314.
30. Treglia G, Castaldi P, Rindi G et al. Diagnostic performance of Gallium-68 somatostatin receptor PET and PET/CT in patients with thoracic and gastroenteropancreatic neuroendocrine tumours: a meta-analysis. Endocrine 2012; 42(1): 80–87. doi: 10.1007/s12020-012-9631-1.
31. Binderup T, Knigge U, Loft A et al. 18F-fluorodeoxyglucose positron emission tomography predicts survival of patients with neuroendo – crine tumors. Clin Cancer Res. 2010; 16: 978e985.
32. Giesel FL, Kratochwil C, Mehndiratta A et al. Comparison of neuroendocrine tumor detection and characterization using DOTATOC-PET in correlation with contrast enhanced CT and delayed contrast enhanced MRI. Eur J Radiol 2012; 81(10): 2820–2825. doi: 10.1016/ j.ejrad.2011.11.007.
33. Dromain C, de Baere T, Lumbroso J et al. Detection of liver metastases from endocrine tumors: a prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. J Clin Oncol 2005; 23(1): 70–78. doi: 10.1200/JCO.2005.01.013.
34. Kunz PL, Reidy-Lagunes D, Anthony LB et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas 2013; 42(4): 557–577. doi: 10.1097/MPA.0b013e31828e34a4.
35. Delle Fave G, Kwekkeboom DJ, Van Cutsem E et al. ENETS consensus guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology 2012; 95(2): 74–87. doi: 10.1159/000335595.
36. Yao JC, Lagunes DR, Kulke MH. Targeted therapies in neuroendocrine tumors (NET): clinical trial challenges and lessons learned. Oncologist 2013; 18(5): 525–532. doi: 10.1634/theoncologist.2012-0434.
37. Wilson MK, Karakasis K, Oza AM. Outcomes and endpoints in trials of cancer treatment: the past, present, and future. Lancet Oncol 2015; 16(1): e32–e42. doi: 10.1016/S1470-2045(14)70375-4.
38. Caplin ME, Pavel M, Cwikla JB et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014; 371(16): 224–233. doi: 10.1056/NEJMc1409757.
39. Rinke A, Muller HH, Schade-Brittinger C et al. Placebo-controlled, double- blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol 2009; 27(28): 4656–4663. doi: 10.1200/JCO.2009.22.8510.
40. Pavel ME, Hainsworth JD, Baudin E et al. Everolimus plus octreotide long- acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo- controlled, phase 3 study. Lancet 2011; 378(9808): 2005–2012. doi: 10.1016/S0140-6736(11)61742-X.
41. Yao JC, Fazio N, Singh S et al. Everolimus in advanced, non-functional neuroendocrine tumors of lung or gastrointestinal origin: efficacy and safety results from the placebo-controlled, double-blind, multicenter, phase 3 study. Lancet 2016; 387(10022): 968–977. doi: 10.1016/S0140-6736(15)00817-X.
42. Rinke A, Wittenberg M, Schade-Brittinger C et al. Placebo controlled, double blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (PROMID): results on long term survival. Neuroendocrinology 2017; 104(1): 26–32. doi: 10.1159/000443612.
43. Yao JC, Fazio N, Singh S et al. Everolimus for the treatment of advanced, non- functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 2016; 387(10022): 968–977. doi: 10.1016/S0140-6736(15)00817-X.
44. Kam BLR, Teunissen JJM, Krenning EP et al. Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging 2012; 39(1): S103–112. doi: 10.1007/s00259-011-2039-y.
45. Strosberg J, Wolin E, Chasen B. 177Lu-Dotatate significantly improves progression-free survival in patients with midgut neuroendocrine tumours: results of the phase III NETTER-1 trial. Pancreas 2016; 45(3): 783. doi: 10.1097/MPA.0000000000000615.
46. Wang J, He A, Feng Q et al. Gastrointestinal mixed adenoneuroendocrine carcinoma: a population level analysis of epidemiological trends. J Transl Med 2020; 18(1): 128. doi: 10.1186/s12967-020-02293-0.
47. Rindi G, Arnold R, Bosman F. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. WHO classification of tumours of the digestive system; 2010: 13–14.
48. La Rosa S, Marando A, Sessa F et al. Mixed adenoneuroendocrine carcinomas (MANECs) of the gastrointestinal tract: an update. Cancers (Basel) 2012; 4(1): 11–30. doi: 10.3390/cancers4010011.
49. Takahashi K, Fujiya M, Sasaki T et al. Endoscopic findings of gastric mixed adenoneuroendocrine carcinoma: a case report. Medicine (Baltimore) 2020; 99(38): e22306. doi: 10.1097/MD.0000000000022306.
50. Lin J, Zhao Y, Zhou Y et al. Comparison of survival and patterns of recurrence in gastric neuroendocrine carcinoma, mixed adenoneuroendocrine carcinoma and adenocarcinoma: A multicenter study from China. JAMA Netw Open 2021; 4(7): e2114180. doi: 10.1001/jamanetworkopen.2021.14180.
51. Rindi G, Bordi C, La RS et al. Gastroenteropancreatic (neuro) endocrine neoplasms: the histology report. Dig Liver Dis 2011; 43(4): S356–S360. doi: 10.1016/S1590-8658(11)60591-4.
52. Tanaka T, Kaneko M, Nozawa H et al. Diagnosis, assessment, and therapeutic strategy for colorectal mixed adenoneuroendocrine carcinoma. Neuroendocrinology 2017; 105(4): 426–434. doi: 10.1159/000478743.
53. Watanabe J, Suwa Y, Ota M et al. Clinicopathological and prognostic evaluations of mixed adenoneuroendocrine carcinoma of the colon and rectum: a case-matched study. Dis Colon Rectum 2016; 59(12): 1160–1167. doi: 10.1097/DCR.0000000000000702.
54. Spada F, Gervaso L, Pisa E et al. Molecular characterization of gastro-entero-pancreatic advanced mixed adeno-neuroendocrine carcinomas: NIRVANA sub-study. Ann Oncol 2022; 33(7): S410–S416. 10.1016/annonc/annonc1060.
55. Xie JW, Lu J, Wang JB et al. Prognostic factors for survival after curative resection of gastric mixed adenoneuroendocrine carcinoma: a series of 80 patients. BMC Cancer 2018; 18(1): 1021. doi: 10.1186/s12885-018-4943-z.
56. Rosa SL, Sessa F, Uccella S. Mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs): unifying the concept of a heterogeneous group of neoplasms. Endocr Pathol 2016; 27(4): 284–311. doi: 10.1007/s12022-016-9432-9.
57. La Rosa S, Marando A, Furlan D et al. Colorectal poorly differentiated neuroendocrine carcinomas and mixed adenoneuroendocrine carcinomas: insights into the diagnostic immunophenotype, assessment of methylation profile, and search for prognostic markers. Am J Surg Pathol 2012; 36(4): 601–611. doi: 10.1097/PAS.0b013e318242e21c.
58. Power DG, Asmis TR, Tang LH et al. High-grade neuroendocrine carcinoma of the colon, long-term survival in advanced disease. Med Oncol 2011; 28(1): S169–S174. doi: 10.1007/s120 32-010-9674-1.
59. Romeo M, Quer A, Tarrats A et al. Appendiceal mixed adenoneuroendocrine carcinomas, a rare entity that can present as a Krukenberg tumor: case report and review of the literature. World J Surg Oncol 2015; 13: 325. doi: 10.1186/s12957-015-0740-1.