Transplantace jater pro PECom napodobující hepatocelulární karcinom
Jana Selucká1, Jiří Froněk Orcid.org 2, Luděk Voska Orcid.org 3, Dana Kautznerová Orcid.org 4, Pavel Taimr Orcid.org 1
+ Pracoviště
Souhrn
PEComy (perivaskulární epiteloidní tumory) jsou poměrně vzácné mezenchymové nádory z perivaskulárních epiteloidních buněk. Jedná se převážně o nádory s benigní biologickou povahou, mohou však mít i maligní potenciál. Vyskytují se v jakékoli anatomické lokalizaci a postihují 4krát častěji ženy většinou mladšího a středního věku. Až 20 % případů je náhodným nálezem na zobrazovacích vyšetřeních. Diagnózu PEComu potvrzuje histologický nález epiteloidních buněk s imunohistochemickým průkazem exprese myoidních a melanocytárních markerů. Jediným kauzálním řešením je chirurgická léčba. Na svízelnost diagnostiky PEComů poukazuje kazuistika pacientky s maligním angiomyolipomem jater, která podstoupila transplantaci jater pro biopticky verifikovanou diagnózu hepatocelulárního karcinomu.
Klíčová slova
perivaskulární epiteloidní neoplazie, angiomyolipom, transplantace jater, hepatocelulární karcinom, mTOR inhibitory
Úvod
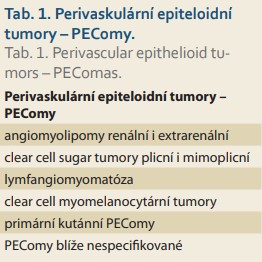
Dle WHO klasifikace jsou PEComy vzácné mezenchymové tumory z perivaskulárních epiteloidních buněk. Do skupiny PEComů patří angiomyolipomy renální i extrarenální (AML), méně známé clear cell sugar tumory plicní i mimoplicní, lymfangiomyomatóza (LAM), clear cell myomelanocytární tumory, primární kutánní PEComy a PEComy blíže nespecifikované (tab. 1).
PEComy postihují nejčastěji ženy mladšího a středního věku, přibližně v poměru 4: 1. Vyskytují se sporadicky nebo asociované s komplexem tuberózní sklerózy (TSC – geneticky podmíněné autozomálně dominantně dědičné onemocnění charakteristické vznikem benigních tumorů postihujících všechny tělesné orgány, nejčastěji kůži, ledviny a CNS; způsobené hereditární nebo nově vzniklou mutací genů TSC1/TSC2).
Nejčastější PEComy a zároveň nejčastější benigní tumory ledvin jsou renální angiomyolipomy s prevalencí v populaci kolem 0,2–0,6 %. Prevalence plicní lymfangiomyomatózy je udávána kolem 3,3–7/1 000 000 žen. Ostatní PEComy jsou popisovány celosvětově v řádu desítek až stovek reportovaných případů. Maligní PEComy jsou extrémně vzácné s incidencí 0,12–0,24/1 000 000 [1]. Renální AML a LAM jsou asociovány s komplexem tuberózní sklerózy v 50 %, resp. 80 % případů [2–4]. U ostatních PEComů byla asociace s TSC popsána v 0–6,25 % případů [5,6].
Vznik PEComů je až v 80 % podmíněn mutací tumor supresorových genů TSC1/TSC2 kódujících proteiny hamartin a tuberin. K alteracím dochází jak v rámci hereditárního přenosu (syndrom TSC), tak i somaticky de novo v průběhu života nemocného. Porucha funkce těchto proteinů vede k nadměrné aktivaci mTOR signální dráhy, a tím i zvýšené buněčné proliferaci. Rizikové faktory pro vznik PEComů nejsou dle dostupných literárních údajů dosud známé.
PEComy se mohou vyskytovat v jakékoli anatomické lokalizaci, avšak nejčastěji byly popsány v orgánech dutiny břišní, retroperitonea a malé pánve. Jsou většinou benigní povahy, nicméně mohou mít i maligní potenciál. Predikce maligního chování na podkladě specifických nálezů zobrazovacích metod nebo histologie není snadná. V klinické praxi se v této souvislosti nejčastěji používá klasifikace dle Folpe et al [5]. Přítomnost dvou a více jím uvedených kritérií (velikost nádoru > 5 cm, infiltrativní růst, přítomnost nekróz, výrazná jaderná pleomorfie, mitotická aktivita > 1/50 HPF) svědčí pro agresivní chování nádoru. Jako nádor s nejasným maligním potenciálem je hodnocen tumor > 5 cm nebo nádor s výrazným jaderným pleomorfizmem či s velkými vícejadernými buňkami. Pokud PEComy metastazují, pak nejčastěji do plic, jater a kostí. Kazuisticky byly rovněž popsány metastázy do CNS, peritonea, lymfatických uzlin, kůže apod. U pacientů s PEComy je nutný nejen komplexní vstupní staging, ale i dlouhodobé sledování po léčbě, neboť PEComy metastazují mnohdy i mnoho let po resekci primárního nádoru [7].
PEComy bývají často asymptomatické a až ve 20 % případů [8] jsou náhodným nálezem na zobrazovacích vyšetřeních, kde ale nemají charakteristický obraz. Proto je definitivní diagnóza PEComu výhradně v rukou patologa a potvrzuje ji přítomnost perivaskulárních epiteloidních buněk.
Perivaskulární epiteloidní buňky (PEC buňky) mají své morfologické a imunohistochemické charakteristiky. Typicky jsou perivaskulárně lokalizované, epiteloidního vzhledu, se světlou až eozinofilní či granulární cytoplazmou, s oválným či kulatým jádrem s nenápadným jadérkem a s žádnými či minimálními buněčnými atypiemi. Předpokládá se, že PEC buňky jsou schopny modulovat svoji morfologii a imunofenotyp, a získat tím fenotyp myoidních buněk či adipocytů, které jsou lokalizovány spíše periferně od cév. Imunohistochemicky je charakteristická exprese myogenních a melanocytárních markerů (HMB-45, melan A, transkripční faktor MITF, desmin, aktin, kaldesmon) [5,6,9–11]. Zajímavostí je, že primární výchozí buňka PEComů nebyla dosud definována.
Ačkoli jsou popsány spíše ojedinělé případy, kdy užití radioterapie či chemoterapie vedlo k redukci nádorové masy nebo stabilizaci choroby, jsou PEComy obecně považovány za chemo- a radiorezistentní a radikální chirurgická resekce je metodou volby. Vzhledem k přítomnosti mutace genů TSC1/TSC2 se využití mTOR inhibitorů při léčbě pokročilých nebo generalizovaných PEComů zdá být nejlepší volbou léčby první linie [12].
Dosud nebyly stanoveny doporučené postupy stran léčby PEComů (vyjma terapie LAM a renálních AML).
Popis případu
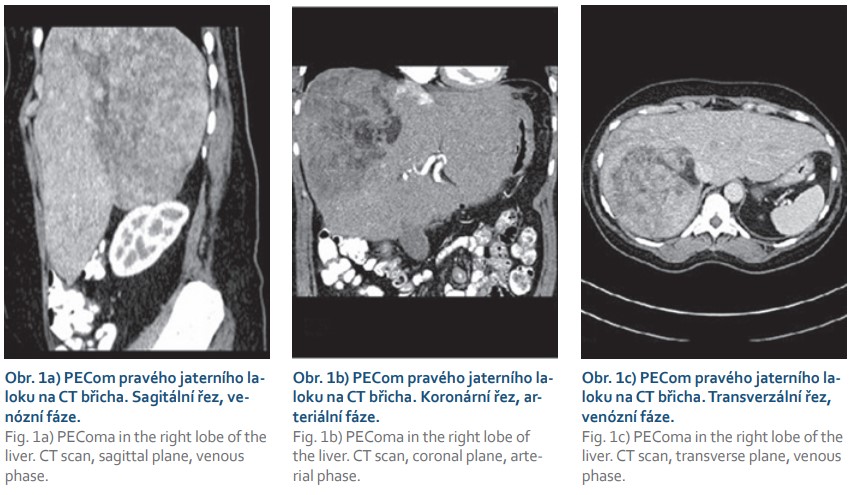
Pacientka (46 let) s arteriální hypertenzí, po appendektomii, byla v květnu 2020 vyšetřována pro bolesti v epigastriu a pravém podžebří. Při fyzikálním vyšetření byla patrná hmatná rezistence v pravém podžebří. Laboratorní nálezy byly kromě lehké trombocytózy v normě včetně jaterních testů a onkomarkerů AFP, CEA, CA 19-9. Sonografie prokázala objemný tumor podjaterní krajiny velikosti 20 cm. CT vyšetření břicha potvrdilo tumor pravého jaterního laloku velikosti 138 × 127 × 200 mm s drobným satelitem v těsné blízkosti (obr. 1a–c), charakterem odpovídajícím hepatocelulárnímu karcinomu. Dále byl popisován uzávěr segmentárních větví portální žíly pro dorzální segmenty pravého jaterního laloku. Levý jaterní lalok byl hypertrofovaný, necirhotického vzhledu. „Shear wave“ elastografie naměřila tuhost z levého laloku 6,9 kPa odpovídající F1 fibróze. Pacientka byla indikována k pravostranné hemihepatektomii. Během operačního výkonu v červnu 2020 byl nález vzhledem k přítomnosti mnohočetných metastatických ložisek (intraoperační sonografie) v obou jaterních lalocích hodnocen jako neresekabilní, peroperační histologie prokázala hepatocelulární karcinom.
V červenci 2020 byla pacientka multidisciplinárním seminářem pro diagnózu hepatocelulárního karcinomu při absenci jaterní cirhózy indikována k transplantaci jater. Byla doplněna protokolární předtransplantační vyšetření, která neprokázala generalizaci maligního onemocnění ani jinou kontraindikaci k transplantaci jater, a pacientka byla zařazena na čekací listinu.
V dalším průběhu pacientka docházela na pravidelné ambulantní kontroly, během nichž byla dominantní potíží bolest břicha reagující vesměs na terapii opioidními analgetiky v kombinaci s paracetamolem. Kontrolní CT břicha v září 2020 popsalo stacionární objemné tumorózní ložisko se satelitem, nebyla nadále patrná portální větev pro dorzální segmenty a periferie pravé jaterní žíly byla zavzata do tumoru. Nález nebyl hodnocen jako makroangioinvaze a pacientka byla ponechána aktivní na čekací listině. Další CT břicha v listopadu 2020 popsalo lehkou progresi velikosti tumoru, nově pak 10mm ložisko levé nadledviny suspektní z metastázy hepatocelulárního karcinomu. Revize předchozích CT vyšetření prokázala přítomnost ložiska již na CT z června 2020, a nález byl tedy hodnocen jako adenom nadledviny.
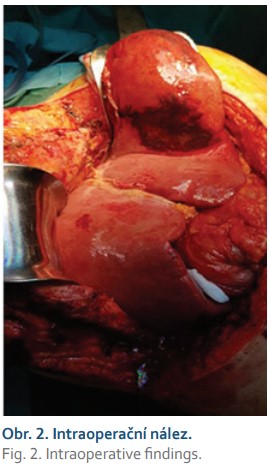
Dne 1. 12. 2020 pacientka podstoupila ortotopickou transplantaci jater s levostrannou adrenalektomií (obr. 2). Výkon byl komplikován oběhovým selháním při reperfuzi štěpu, následně byla provedena první operační revize pro hemoperitoneum s ošetřením zdroje krvácení, druhá operační revize následovala pro podezření na poruchu perfuze jaterního štěpu při významné elevaci jaterních testů, která však nebyla prokázána. Byla indikována podpůrná léčba (CRRT – kontinuální náhrada funkce ledvin, a FPSA – frakcionovaná plazmatická separace a adsorpce), po které následovalo zlepšení jaterních parametrů. Průběh hospitalizace byl komplikován rozvojem deliria. Po stabilizaci stavu byla pacientka přeložena na chirurgický JIP a následně standartní oddělení, kde byla léčena pravostranná bronchopneumonie a titrována hladina imunosuprese. Dne 22. 12. 2020 byla pacientka propuštěna do ambulantní péče.
Definitivní histologický nález z explantátu prokázal 150mm a 15mm ložisko infiltrativně rostoucího epiteloidního angiomyolipomu jater (PEComu) nejistého maligního potenciálu. Dle Folpeho kritérií se jednalo o maligní epiteloidní angiomyolipom. V resekátu levé nadleviny byla zastižena mikronodulární hyperplazie.
Pacientka nadále podstupuje pravidelná restagingová vyšetření, jejichž výsledky zatím prokazují remisi.
Diskuze
Angiomyolipomy jater patří do skupiny vzácných mezenchymových tumorů, tzv. PEComů, charakterizovaných přítomností perivaskulárních epiteloidních buněk. Zatímco renální angiomyolipom je poměrně známý, jaterní angiomyolipom se vyskytuje vzácně. První jaterní angiomyolipom popsali ve své práci Ishak et al v roce 1976 [13] a do roku 2017 bylo popsáno přibližně dalších 600 případů [2].
Angiomyolipomy jater se vyskytují nejčastěji v necirhotických játrech a stejně jako ostatní PEComy postihují převážně ženy mladšího a středního věku [2]. Z celé rodiny PEComů jsou právě angiomyolipomy a lymfangiomyomatóza nejčastěji asociovány s komplexem tuberózní sklerózy (TSC). Zatímco až 50 % renálních angiomyolipomů je spojeno s TSC, v případě pacientů s jaterním angiomyolipomem je asociace s TSC popsána v 5–15 % [2,3].
Jedná se většinou o benigní asymptomatické léze. Symptomatické angiomyolipomy se projevují nejčastěji bolestmi břicha a hmotnostním úbytkem. Mohou mít maligní potenciál a byly dokonce popsány i případy spontánní ruptury. Riziko maligního zvratu u angiomyolipomů jater je kolem 4,1 % a celková mortalita spojená s jaterním angiomyolipomem se pohybuje kolem 0,8 % [2].
Angiomyolipomy jater jsou většinou náhodným nálezem na zobrazovacích vyšetřeních (42–72 %) [2,14,15]. Na CT a MR se zobrazují jako solidní hypervaskularizovaná ložiska s obsahem tuku a wash-outem a mohou být velmi snadno zaměněny například s hepatocelulárním karcinomem. Dle systematické review z roku 2017 byla správná diagnóza angiomyolipomu jater pomocí zobrazovacích metod určena ve 28,2 % [2].
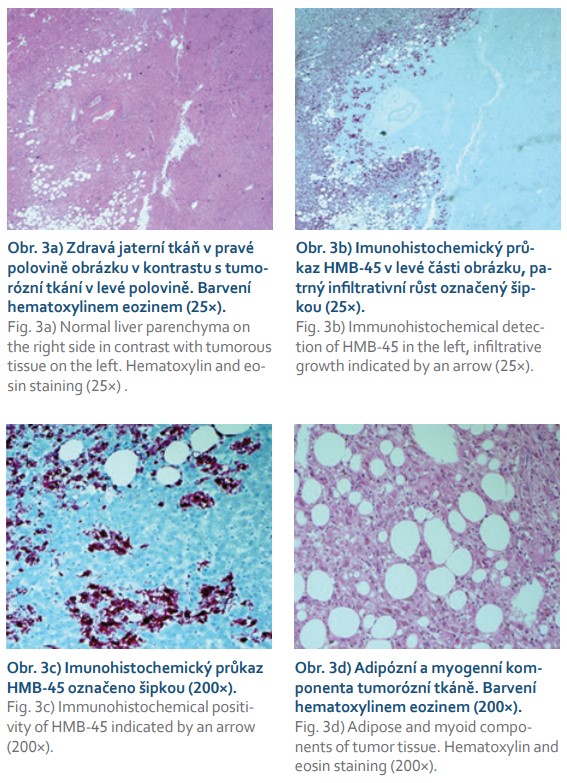
Histologicky je typická přítomnost perivaskulárně lokalizovaných epiteloidních buněk, tukové buňky a vřetenovité buňky připomínající buňky hladké svaloviny jsou lokalizovány periferněji od cév. Dle zastoupení jednotlivých složek lze angiomyolipomy dělit na konvenční angiomyolipomy s převahou cév, lipomatózní nebo myomatózní tkáně, a epiteloidní angiomyolipomy, kde je zastoupení epiteloidních buněk v 10–100 %. Bylo popsáno 14 případů zánětlivého angiomyolipomu jater, pro který je charakteristická infiltrace zánětlivými buňkami (lymfocyty, plazmocyty, histiocyty) [16]. Epiteloidnímu typu angiomyolipomu je přičítán nejvyšší maligní potenciál [14]. Imunohistochemicky je pro jaterní angiomyolipom typická exprese melanocytárních markerů (HMB-45 a melan A) a markerů hladké svaloviny (aktin a/nebo desmin), přičemž nejspecifičtějším markerem je právě HMB-45, který Klompenhouwerová et al prokázali u 91,5 % pacientů [2]. Ve shodě s literárními údaji nalezli patologové i v jaterním explantátu naší pacientky pozitivitu HMB-45, melanu A a aktinu (obr. 3a–d), imunohistochemické vyšetření nebylo provedeno při předchozí peroperační biopsii, neboť se standardně peroperačně neprovádí. Jak dokazuje naše kazuistika, je použití imunohistochemických metod pro diagnostiku angiomyolipomu jater nezbytné, neboť samotný histologický nález je velmi obtížně odlišitelný např. od hepatocelulárního karcinomu. V rámci histologické diferenciální diagnostiky je třeba dále vyloučit konvenční melanom, sarkom z jasných buněk, gastrointestinální stromální tumory, nádory vycházející ze svalové či tukové tkáně. Dle multicentrické retrospektivní analýzy publikované v roce 2020 byla správná preoperační histologická diagnóza stanovena v 84 % případů [14].
Léčba jaterního angiomyolipomu by měla vycházet ze znalosti histologie a multidisciplinárního přístupu. Za jedinou kurativní léčbu je považována léčba chirurgická. Podstoupilo ji více než 75 % pacientů s angiomyolipomem jater [2,14]. Chirurgická léčba je indikována v případě symptomatického tumoru, při nejasném histologickém nálezu, progresi velikosti tumoru či při velikosti tumoru > 5 cm. Rekurence po chirurgické resekci je udávána kolem 2,4 % [2], v případě epiteloidního angiomyolipomu jater se postresekční lokální či vzdálená rekurence pohybuje kolem 10 % [17].
Byly popsány i případy využití jiných léčebných modalit jako např. užití radiofrekvenční ablace nebo chemoembolizace, v případě lézí s předpokládanou benigní biologickou povahou byla zvolena i radiologická dispenzarizace. Optimální intervaly radiologického sledování ale nejsou jednoznačně stanoveny.
Transplantace jater pro angiomyolipom jsou raritní. První dokumentovaná transplantace jater byla dle dostupné literatury provedena v roce 2010 a od té doby následovalo ještě několik málo případů, včetně dalšího případu pacientky s angiomyolipomem jater transplantované v IKEM v lednu 2021. Většina případů byla primárně indikována z důvodů velikosti či množství jaterních ložisek, v jednom případě byl tumor jater považován za hemangiosarkom. Jediná větší multicentrická studie [14] vycházející z dat šesti světových transplantačních center z let 1997–2017 byla publikována v roce 2020. V souboru byli nalezeni pouze tři pacienti s jaterním angiomyolipomem, kteří podstoupili transplantaci jater. Je však třeba zdůraznit, že ve všech těchto případech byl angiomyolipom překvapujícím pooperačním nálezem v explantátu jater. Skutečnost, že u těchto nemocných byla indikací k transplantaci jater diagnóza hepatocelulárního karcinomu stanovená 2krát dle rentgenologických kritérií a jednou po bioptické verifikaci, ukazuje na svízelnost stanovení správné diagnózy angiomyolipomu v bioptických vzorcích.
Farmakologická léčba je určena k terapii neoperabilních a generalizovaných angiomyolipomů jater a ve své podstatě není ani přesně definována. Chemoterapie se ukázala jako málo efektivní. V terapii angiomyolipomů ledvin a TSC jsou běžně užívány inhibitory mTOR, v případě jaterních angiomyolipomů jsou popsány spíše jednotlivé kazuistiky, ve kterých užití mTOR inhibitorů v léčbě jaterních angiomyolipomů asociovaných s komplexem tuberózní sklerózy vedlo k redukci nádorové masy [18]. Podobný efekt byl popsán i u použití tamoxifenu v terapii jaterního angiomyolipomu s expresí estrogenových a progesteronových receptorů u pacientky s TSC [19].
Závěr
Popisujeme případ pacientky, která pro diagnózu hepatocelulárního karcinomu podstoupila transplantaci jater. V explantátu však byl prokázán epiteloidní angiomyolipom, poměrně vzácný zástupce rodiny PEComů. Angiomyolipomy jater jsou vzhledem k variabilitě histologických nálezů i nálezů na zobrazovacích vyšetřeních pro patology i radiology diagnostickou výzvou. Dosud neexistují histologická ani radiologická kritéria, která by jednoznačně určila jejich biologickou povahu. Terapie těchto tumorů vyžaduje multidisciplinární přístup. Transplantace je jednou z možností ošetření, diagnóza nemusí být v době indikace, resp. transplantace známa, tak jako v našem případě.
ORCID autorů
J. Froněk ORCID 0000-0003-2379-3886,
L. Voska ORCID 0000-0001-6281-8973,
D. Kautznerová ORCID 0000-0002-1238-2451,
P. Taimr ORCID 0000-0002-2655-7603.
Doručeno/Submitted: 1. 3. 2022
Přijato/Accepted: 23. 3. 2022
MUDr. Jana Selucká
Klinika hepatogastroenterologie, IKEM
Vídeňská 1958/9
140 21 Praha 4
jana.selucka@ikem.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Sobiborowicz A, Świtaj T, Teterycz P et al. Feasibility and long-term efficacy of PEComa treatment – 20 years of Experience. J Clin Med 2021; 10(10): 2200. doi: 10.3390/jcm10102 200.
2. Klompenhouwer AJ, Verver D, Janki S et al. Management of hepatic angiomyolipoma: a systematic review. Liver Int 2017; 37(9): 1272–1280. doi: 10.1111/liv.13381.
3. Kamimura K, Nomoto M, Aoyagi Y. Hepatic angiomyolipoma: diagnostic findings and management. Int J Hepatol 2012; 2012: 410781. doi: 10.1155/2012/410781.
4. Cudzilo CJ, Szczesniak RD, Brody AS et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest 2013; 144(2): 578–585. doi: 10.1378/chest.12-2813.
5. Folpe AL, Mentzel T, Lehr HA et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 2005; 29(12): 1558–1575. doi: 10.1097/01.pas.0000173232.22117.37.
6. Schoolmeester JK, Howitt BE, Hirsch MS et al. Perivascular epithelioid cell neoplasm (PEComa) of the gynecologic tract: clinicopathologic and immunohistochemical characterization of 16 cases. Am J Surg Pathol 2014; 38(2): 176–188. doi: 10.1097/PAS.0000000000000133.
7. Parfitt JR, Bella AJ, Izawa JI et al. Malignant neoplasm of perivascular epithelioid cells of the liver. Arch Pathol Lab Med 2006; 130(8): 1219–1222. doi: 10.5858/2006-130-1219-MNO PEC.
8. Zizzo M, Ugoletti L, Tumiati D et al. Primary pancreatic perivascular epithelioid cell tumor (PEComa): a surgical enigma. A systematic review of the literature. Pancreatology 2018; 18(3): 238–245. doi: 10.1016/j.pan.2018.02.007.
9. Bennett JA, Braga AC, Pinto A et al. Uterine PEComas: a morphologic, immunohistochemical, and molecular analysis of 32 tumors. Am J Surg Pathol 2018; 42(10): 1370–1383. doi: 10.1097/PAS.0000000000001119.
10. Hornick JL, Fletcher CD. Sclerosing PEComa: clinicopathologic analysis of a distinctive variant with a predilection for the retroperitoneum. Am J Surg Pathol 2008; 32(4): 493–501. doi: 10.1097/PAS.0b013e318161dc34.
11. Armah HB, Parwani AV. Malignant perivascular epithelioid cell tumor (PEComa) of the uterus with late renal and pulmonary metastases: a case report with review of the literature. Diagn Pathol 2007; 2: 45. doi: 10.1186/1746-1596-2-45.
12. Świtaj T, Sobiborowicz A, Teterycz P et al. Efficacy of Sirolimus treatment in PEComa-10 years of practice perspective. J Clin Med 2021; 10(16): 3705. doi: 10.3390/jcm10163705.
13. Ishak KG. Mesenchymal tumors of the liver. In: Okuda K, Peters RL (eds). Hepatocellular carcinoma. NY: John Wiley and Sons 1976: 247–307.
14. Klompenhouwer AJ, Dwarkasing RS, Doukas M et al. Hepatic angiomyolipoma: an international multicenter analysis on diagnosis, management and outcome. HPB (Oxford) 2020; 22(4): 622–629. doi: 10.1016/j.hpb.2019.09.004.
15. Yang X, Lei C, Qiu Y et al. Selecting a suitable surgical treatment for hepatic angiomyolipoma: a retrospective analysis of 92 cases. ANZ J Surg 2018; 88(9): E664–E669. doi: 10.1111/ans.14323.
16. Mao JX, Yuan H, Sun KY et al. Pooled analysis of hepatic inflammatory angiomyolipoma. Clin Res Hepatol Gastroenterol 2020; 44(6): e145–e151. doi: 10.1016/j.clinre.2020.04.005.
17. Liu J, Zhang CW, Hong DF et al. Primary hepatic epithelioid angiomyolipoma: a malignant potential tumor which should be recognized. World J Gastroenterol 2016; 22(20): 4908–4917. doi: 10.3748/wjg.v22.i20.4908.
18. Dabora SL, Franz DN, Ashwal S et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF- D levels decrease. PLoS One 2011; 6(9): e23379. doi: 10.1371/journal.pone.0023379.
19. Lenci I, Angelico M, Tisone G. Massive hepatic angiomyolipoma in a young woman with tuberous sclerosis complex: significant clinical improvement during tamoxifen treatment. J Hepatol 2008; 48(6): 1026–1029. doi: 10.1016/j.jhep.2008.01.036.