Wilsonova choroba v súbore detských pacientov na Slovensku
Jana Kosnáčová1, Ľudmila Podracká1
+ Pracoviště
Souhrn
Úvod: Wilsonova choroba (WD – Wilson’s disease) je zriedkavé, metabolické, geneticky podmienené ochorenie. Pacienti môžu byť rôzne dlho asymptomatickí alebo sa ochorenie manifestuje nešpecifickými príznakmi až po orgánovo špecifické postihnutie. Diagnostika sa opiera o klinický obraz a analýzu cielených výsledkov. Včas diagnostikované ochorenie je liečiteľné, základnou terapiou je chelatačná liečba. Pacienti s hepatálnym zlyhaním sú indikovaní na transplantáciu pečene. Metódy a súbor: Autori retrospektívne hodnotia súbor 17 pacientov s WD diagnostikovanou v detskom a adolescentnom veku. Výsledky: V priebehu rokov 2000–2015 na I. detskej klinike LF UK a DFNsP v Bratislave bola WD potvrdená u 17 pacientov od 3. do 18. roku života. Najčastejšie sa vyskytovala hepatálna forma WD, a to u 13 pacientov (77 %). U dvoch pacientov sa WD manifestovala akútnym zlyhaním pečene v adolescentnom veku. Deväť pacientov malo dlhodobo zvýšenú aktivitu aminotransferáz. U dvoch pacientov sme identifikovali WD až v štádiu cirhózy. Neurologická symptomatológia dominovala u dvoch pacientov. Najčastejšiu genetickú mutáciu v stredoeurópskom regióne H1069Q sme potvrdili u 6 homozygotných nosičov, 10 pacienti boli zložení heterozygoti. U jedného pacienta sa nepodarilo ani rozšíreným genetickým vyšetrením potvrdiť patologickú mutáciu. Tri deti z kohorty podstúpili transplantáciu pečene, 2 asymptomatické deti sú na liečbe zinkom, 11 pacientov je liečených penicilamínom a 1 dieťa trientine dihydrochloridom. Záver: WD je zriedkavým ochorením detského veku. Hepatálna forma je najčastejšou manifestáciou, zvyčajne sa prejaví v druhom decéniu. Stanovenie diagnózy je spojené s úskaliami, neexistuje jediný vysoko senzitívny či špecifický parameter na jednoznačné určenie diagnózy. Screeningovým vyšetrením je koncentrácia ceruloplazmínu v sére. Pomocným vyšetrením je plazmatická hladina a renálna exkrécia medi. Zlatým štandardom je stanovenie obsahu medi v sušine pečene. Prínosom pre potvrdenie ochorenia je genetická diagnostika, najmä u prvostupňových príbuzných, asymptomatických jedincov. Včasná diagnostika a liečba môže zabrániť progresii ochorenia. Bez lie?by je ochorenčby je ochorenie fatálne.
Klíčová slova
Wilsonova choroba, ceruloplazmin, měď, měď v sušiněÚvod
Wilsonova choroba (WD – Wilson’s disease; hepatolentikulárna degenerácia, McKusick č. 277 900) je vrodené, geneticky podmienené metabolické ochorenie s autozómovo-recesívnym typom dedičnosti [1]. S prevalenciou 1: 30 000 živonarodených detí (variácia 1: 18 000 až 1: 700 000) patrí medzi zriedkavé ochorenia. Gén ATP7B pre WD je lokalizovaný na dlhom ramienku 13. chromozómu v oblasti 13q 14.3 a bol identifikovaný roku 1993 [2]. Gén ATP7B kóduje meď (Cu) transportujúcu ATPázu typu P zodpovednú za metabolizmus, transport a exkréciu Cu do žlče. Ak je gén ATP7B defektný, Cu z potravy sa nemôže inkorporovať do ceruloplazmínu ani vylúčiť exkréciou do žlče a postupne sa hromadí najmä v pečeni, mozgu, rohovke a v obličkách, ktoré toxicky poškodzuje. Gén ATP7B sa vyznačuje veľkou variabilitou. Doteraz bolo identifikovaných viac ako 800 kauzálnych mutácií [2]. Najčastejšou mutáciou v strednej Európe je zámena histidínu za glutamín v pozícii 1069 (H1069Q), resp. p.His1069Gln, ktorú v tejto geografickej oblasti má 30–60 % pacientov kaukazskej rasy [3–5]. Výskyt heterozygotných nosičov génu sa udáva 1: 90 obyvateľov [6–8]. Pri suspekcii na WD sa vykonáva genotypová analýza najčastejších mutácií. Pozitívna genetika potvrdzuje ochorenie, negatívny výsledok informuje o neprítomnosti vyšetrovaných mutácií, ale ochorenie nevylučuje.
Klinický obraz WD je veľmi pestrý. Ochorenie sa môže prejaviť v ktoromkoľvek veku, po rôzne dlhej dobe latencie. Prvé príznaky sa najčastejšie manifestujú vo veku 5–35 rokov [9]. Podľa dominantných klinických prejavov sa rozlišuje forma hepatálna, neurologická a psychiatrická (neurologicko-psychiatrická). Pri rozvinutom ochorení sa kombinujú viaceré klinické či orgánové symptómy. Je zaujímavé, že aj v rámci jednej rodiny môžu mať postihnutí jedinci rôznorodé príznaky [10,11]. Hepatálna forma je charakteristická manifestácia WD detského a adolescentného veku. Postihnutie pečene sa môže prejaviť zvýšenou aktivitou aminotransferáz, akútnou hepatitídou, steatózou pečene, chronickým ochorením pečene, ale aj akútnym zlyhaním pečene [12,13], ktoré môže byť dokonca prvým prejavom WD. Ochorenie prebieha najčastejšie plíživo s pomalou progresiou a môže sa diagnostikovať až v štádiu cirhotickej prestavby pečene.
U adolescentov a mladých dospelých sa WD môže prejaviť variabilnou neurologickou symptomatológiou. Ochorenie začína nenápadne zmenou správania a nálady, zmenou intelektových funkcií, zhoršením študijných a pracovných výsledkov, tremorom, dyzartriou, grimasovaním, poruchami chôdze, orolingválnymi dyskinézami, dystóniou a mozočkovými príznakmi [14]. Neurologické symptómy sú viac alebo menej sprevádzané psychiatrickými príznakmi a charakteristickými oftalmologickým nálezmi ako je Kayserov-Fleischerov prstenec alebo slnečnicová katarakta. K menej častým prejavom WD patrí renálne postihnutie, kostné a endokrinologické zmeny. Ataky hemolytickej anémie s negatívnymi Coombsovými testami sú u detí s WD relatívne časté, môžu sa pridružiť pri akútnom zlyhaní pečene a/alebo pri nasadení liečby. Diagnostika sa opiera o klinický obraz, biochemické, histologické, oftalmologické, genetické a neurologické vyšetrenie. Základným diagnostickým vyšetrením pre WD je vyšetrenie sérovej koncentrácie ceruloplazmínu, pomocným vyšetrením je hladina sérovej Cu a exkrécia Cu močom. Za diagnosticky najspoľahlivejší ukazovateľ sa považuje dôkaz obsahu Cu v sušine pečene. Diagnostika asymptomatických pacientov je náročná, keďže pozitivitu laboratórnych markerov zistíme náhodne pri vyšetrení pre iné ochorenie alebo cieleným vyšetrením bezpríznakových prvostupňových príbuzných, avšak signifikantne býva zvýšený obsah Cu v sušine [9]. Molekulovo-genetická analýza v súčasnosti je rutinná vyšetrovacia metóda a významne prispieva k diagnostike WD. V súčasnosti je známych až 800 rozličných mutácií [2]. Treba však podčiarknuť, že negatívny výsledok molekulovo-genetickej analýzy nevylučuje prítomnosť ochorenia, informuje len o neprítomnosti vyšetrovaných mutácií, lebo pacient môže byť nosičom inej genetickej mutácie. Genetické testovanie a poradenstvo sa odporúča u súrodencov a prvostupňových príbuzných [15]. K diagnóze WD môže prispieť tiež neurológ či psychiater, magnetická rezonancia (MR) mozgu môže preukázať ukladanie Cu v mozgu.
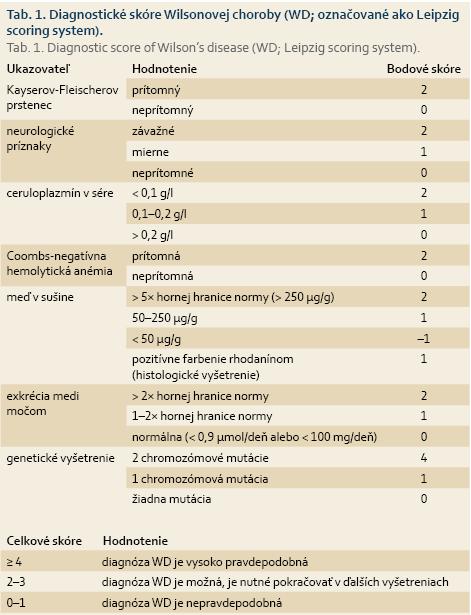
Vzhľadom na rozmanitosť klinického obrazu a nešpecifické biochemické parametre, na diagnostiku WD sa využíva Diagnostické skóre WD [12].
V skórovacom systéme sa hodnotí: 1. Kayserov-Fleischerov prstenec, 2. neurologická symptomatológia, 3. ceruloplazmín v sére, 4. hemolytická anémia s negatívnym Coombsovým testom, 5. obsah Cu v pečeňovej sušine, 6. močová exkrécia Cu, 7. mutačná analýza (tab. 1).
Prítomnosť viac ako 4 bodov zo škály skórovacích kritérií potvrdzuje diagnózu WD [3,12,16]. Asymptomatickí pacienti v mladších vekových kategóriách často unikajú diagnóze [17]. Včasná diagnóza a terapia môžu zastaviť progresiu ochorenia.
Lie?ebn? strat?gčebné stratégie zahŕňajú predovšetkým medikamentóznu liečbu chelátormi ako je D-penicilamín a trientine dihydrochloride. V liečbe sa využíva aj zinok, ktorý ovplyvňuje vstrebávanie Cu v tráviacom trakte. Samotná diéta s nízkym obsahom Cu nemá dostatočný terapeutický efekt, má doplnkový význam v úvode liečby. Najpoužívanejším chelátotvorným liekom je D-penicilamín, ktorý viaže Cu a vytvára komplexy vylučované močom. Transplantácia pečene je indikovaná pri progredujúcom akútnom hepatálnom zlyhaní a dekompenzácii chronického ochorenia pečene a je jedinou možnou liečbou fulminantnej formy WD.
Longitudinálne klinické dáta u chorých s WD sú pre zriedkavý výskyt limitované. Údaje o frekvencii a priebehu WD u detí na Slovensku nie sú známe. V našej retrospektívnej štúdii analyzujeme súbor 17 slovenských pediatrických pacientov s WD.
Klinický súbor a výsledky
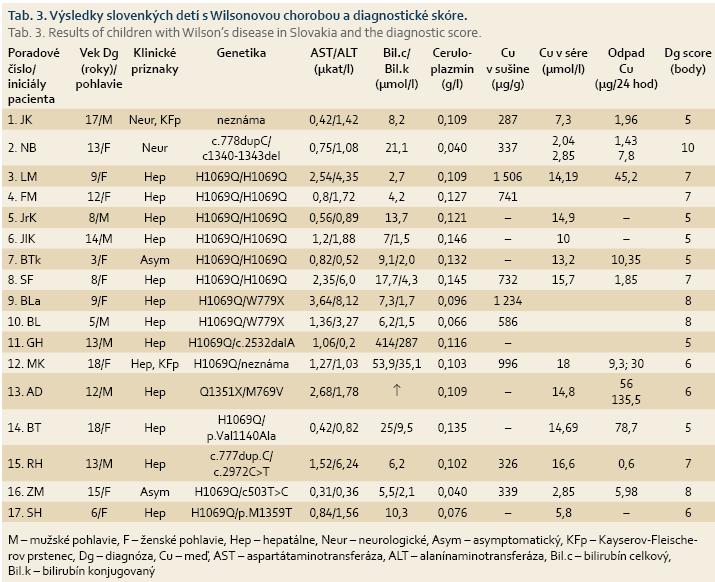
Na 1. detskej klinike LF UK a DFNsP v Bratislave sme v priebehu rokov 2000 až 2015 diagnostikovali WD u 17 detí nepríbuzenských rodičov, z toho u 10 (58,8 %) dievčat. Priemerný vek diagnózy ochorenia v súbore je 11,35 roka ± 4,48 rokov (3–18 rokov). Pri diagnostike WD sme sa opierali o Diagnostické skóre WD (tab. 1). Za vysoko pravdepodobnú WD sme považovali skóre viac ako 4 body. Ceruloplazmín sme vyšetrovali imunoturbidimetricky na prístroji COBAS INTEGRA firmy Roche. Cu v sére a v moči sme vyšetrovali plameňovou atómovou absorpčnou spektrometriou. Obsah Cu v sušine sa stanovoval v Hepatologickom laboratóriu VFN v Prahe. Molekulovo-genetickú analýzu sme vykonali na Ústave lekárskej biológie a klinickej genetiky LF UK a UN v Bratislave a rozšírenú diagnostiku na Oddelení lekárskej genetiky MU v Brne pod vedením doc. MUDr. D. Procházkovej, CSc.
Výsledky
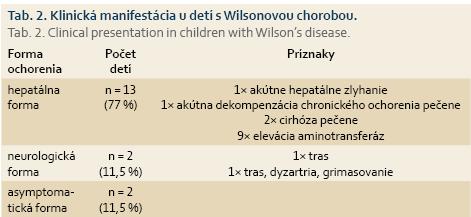
U 7 detí sa WD zistila v prvej dekáde života, u 10 pacientov v druhej dekáde, najmladší pacient mal v čase diagnózy iba 3 roky. U štyroch (23 %) chorých sa WD vyskytovala v príbuzenstve – u súrodencov. V úvodných klinických prejavoch dominovala hepatálna symptomatológia (13 detí, 77 % z celého súboru), 2 pacienti mali charakteristické neurologické príznaky a 2 deti boli asymptomatické (tab. 2). Stupeň poškodenia pečene interindividuálne varíroval, od akútneho hepatálneho zlyhania u jedného dieťaťa až po náhlu dekompenzáciu chronického, dovtedy bližšie „nešpecifikovaného ochorenia pečene“ (jeden pacient). U dvoch dievčat sa WD potvrdila v štádiu cirhózy pečene, u deviatich detí sme zachytili zvýšenú aktivitu aminotransferáz alebo subklinickú hepatitídu. Iniciálnymi neurologickými príznakmi (tras horných končatín, dyzartria a grimasovanie) sa ochorenie manifestovalo u dvoch pacientov. Ani jeden z pacientov s neurologickou symptomatológiou nemal potvrdenú cirhózu pečene. Typický Kayserov-Fleischerov prstenec mal chlapec s neurologickou formou a dievča s pokročilou cirhózou pečene.
Biochemické parametre u jednotlivých chorých preukazovali značnú variáciu. Zníženú koncentráciu ceruloplazmínu v sére – pod 0,15 g/l (od 0,04 do 0,146 g/l) sme potvrdili u všetkých 17 chorých. Vyššiu aktivitu aminotransferáz mali 14 pacienti, a 1/3 aj hyperbilirubinémiu. Cu v sére bola znížená u 5 pacientov, signifikantne vyššiu bazálnu renálnu exkréciu Cu mal len 1 pacient s akútnym zlyhaním pečene (tab. 3). Biopsiu pečene sme vykonali u 10 pacientov pred nasadením liečby. V histologickom obraze boli prítomné nešpecifické zmeny ako je minimálny portálny zápal bez signifikantnej „interface“ aktivity, bez nekróz, minimálny lobulárny zápal, ľahká fibróza, bez deplécie žlčovodov, prípadne len mierna duktálna reakcia, stupeň steatózy v histologickom materiáli sa pohyboval od 5 do 80 %. Špeciálne farbenie na železo, Cu a žlčové farbivá bolo negatívne. U všetkých bioptovaných sa potvrdil zvýšený obsah Cu v sušine pečene (> 250 μg Cu/g sušiny, norma do 50 μg Cu/g sušine).
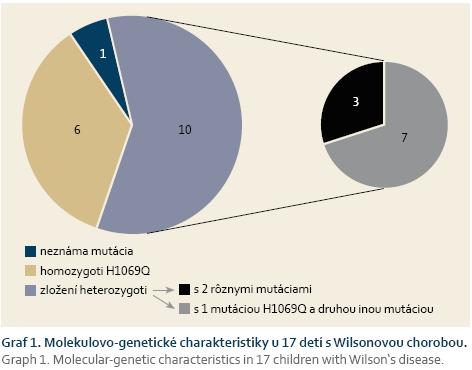
DNA analýza sa realizovala u všetkých 17 detí. U 16 pacientov sme identifikovali kauzálne mutácie, len u 1 probanda sa patologická mutácia vo vybranom spektre známych mutácií nedetegovala (graf 1). Šiesti pacienti (35 %) sú homozygotní nosiči najfrekvenčnejšej mutácie H1069Q v stredoeurópskom regióne; 10 probandi sú zložení heterozygoti (tab. 3). Z 10 zložených heterozygotov 7 pacienti majú jednu alelu H1069Q a druhú inú patologickú alelu.
V našej kohorte sme zistili širokú genotypovo-fenotypovú variáciu patognomických nálezov. Prejavy ochorenia sa líšili aj u probandov s rovnakým genotypom (tab. 2). U adolescenta s neurologickou manifestáciou sa v testovanom genetickom paneli nenašli žiadne mutácie ATP7B génu, ale WD sme dokázali podľa vysokého obsahu Cu v sušine a MR nálezu kumulácie Cu v bazálnych gangliách.
Liečbu D-penicilamínom sme indikovali u 12 pacientov. V úvode sme podávali 2–5 mg/kg a postupne sme dávku každý týždeň zvyšovali až na maximum 20 mg/kg/deň rozdelenú do dvoch dávok. Celkom 11 pacientov toleruje liečbu D-penicilamínom dobre, pacientov sledujeme až do dosiahnutia veku 18 rokov. U jedného dieťaťa sme pre závažný nežiaduci účinok penicilamínu (status dystonicus) zmenili terapiu na trientine dihydrochloride. Ide o prvého pacienta liečeného trientine dihydrochloridom na Slovensku. Terapia je efektívna a neurologická symptomatológia ustúpila. Úspešná transplantácia pečene bola vykonaná u troch detí s progresívnou hepatálnou formou WD. Transplantácia pečene bola vykonaná 1× pre akútne zlyhanie pečene, ktoré bolo prvou manifestáciou dovtedy nemanifestovaného ochorenia u pacienta vo veku 12 rokov. Ďalší pacient sa transplantoval vo veku 13 rokov pre akútnu dekompenzáciu chronického ochorenia nejasnej etiológie, ktoré nebolo v priebehu niekoľkých rokov dovyšetrované pre nespoluprácu rodiny a v jednom prípade sme transplantáciu indikovali pre cirhózu pečene zistenú v 18. roku života [18]. Dvaja asymptomatickí pacienti sú dlho-dobo na monoterapii zinkom.
Diskusia
Longitudinálne údaje o WD u detí v stredoeurópskom regióne sú limitované. Prvé súhrnné údaje publikovali Procházková et al roku 2005 v súbore 22 českých detí [19]. Najpočetnejšiu pediatrickú kohortu z centrálnej Európy reprezentuje súbor 156 detí z terciárnych pediatrických gastroenterologických centier v Poľsku [3]. Naša retrospektívna štúdia predstavuje prvú kohortu pediatrických pacientov s WD na Slovensku, ktorá analyzovala klinické prejavy, biochemické a genetické testy. Celkom 58 % našich chorých malo v čase diagnostiky viac ako 10 rokov, častejší výskyt sme potvrdili u dievčat (58,8 %). Z orgánových prejavov dominovala hepatálna forma (77 %), iba u dvoch adolescentov sa WD prejavila iniciálnou neurologickou symptomatológiou. Klinické dáta u slovenských detí s WD korešpondujú s českou a poľskou kohortou. U 9 z 22 českých detí sa ochorenie prejavilo v prvej dekáde života, a len 13 z celého počtu 156 chorých v poľskej kohorte bolo mladších ako 5 rokov [3,19]. Diagnostika WD môže byť zložitá, lebo klinická symptomatológia je rozmanitá a v súčasnosti neexistuje jeden špecifický test per se, ktorý potvrdzuje WD. V lekárskej praxi môžu byť nápomocné skórovacie systémy, ktoré využívajú kombináciu klinických kritérií a laboratórnych testov [3,12,16,20]. Na úskalia diagnostiky poukazujú heterogénne patologické nálezy medzi jednotlivcami v našom súbore. U detí je diagnostika zložitejšia, lebo charakteristické očné príznaky a neurologické prejavy majú prolongovanú latenciu a vyskytujú sa podstatne zriedkavejšie ako u dospelých pacientov. V našej kohorte mali priekazný Kayserov-Fleischerov prstenec dvaja pacienti vo veku 13 a 17 rokov, z nich jedno dieťa malo neurologické symptómy. Znížený ceruloplazmín v sére je základný ale nešpecifický marker, lebo až 20 % detí a dospelých s WD má normálne koncentrácie ceruloplazmínu [12,20]. Nízky ceruloplazmín môže sprevádzať aj iné ochorenia (napr. nefrotický syndróm, autoimunitná hepatitída, pokročilé pečeňové zlyhanie, celiakia) a selektovať heterozygotov, ktorí nevykazujú známky kumulácie Cu [12]. Pri interpretácií výsledkov treba pamätať, že ceruloplazmín je proteín akútnej fázy a môže stúpať pri systémovom a/alebo lokalizovanom zápale v organizme. Pre tieto obmedzenia sa na dôkaz poruchy metabolizmu Cu odporúča kombinácia biochemických testov. Všetci naši pacienti mali nízky ceruloplazmín v sére, ale bazálna exkrécia Cu interindividuálne varírovala. Signifikantne vyššiu bazálnu exkréciu Cu sme zistili len u jedného pacienta s akútnym zlyhaním pečene. Podobne Procházková et al udávajú vysoký bazálny odpad Cu v moči len u 18 % detí [19]. V poľskej kohorte malo 90,24 % detí nízky ceruloplazmín a 51,93 % bazálnu močovú exkréciu Cu > 100 μg/24 hod [3].
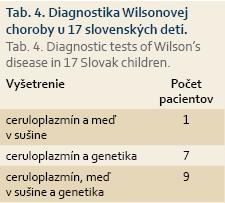
Veľkým prínosom pre diagnostiku WD je molekulárno-genetické vyšetrenie. Z doteraz opísaných viac ako 800 rozličných mutácií ATP7B génu sa v centrálnej Európe najčastejšie vyskytuje mutácia H1069Q s frekvenciou 30–60 % v jednotlivých sériách pacientov z Poľska, Rumunska, Rakúska a ďalších európskych krajín. Podľa očakávania má slovenská kohorta porovnateľnú distribúciu genotypov, 35 % pacientov sú homozygotní nosiči mutácie H1069Q, 10 probandi sú zložení heterozygoti. Detekcia mutácií na obidvoch chromozómoch potvrdzuje WD, ale chýbanie mutácie ochorenie nevylučuje. Názorne to dokumentuje komplikovaný prípad nášho pacienta s iniciálnou neurologickou symptomatológiou, u ktorého sme v testovanom genetickom paneli neidentifikovali žiadne mutácie ATP7B génu. WD sme stanovili podľa nízkej sérovej hodnoty ceruloplazmínu a vysokého obsahu Cu v sušine. Diagnózu podporil aj oftalmologický nález Kayserovho-Fleischerovho prstenca a charakteristický obraz MR mozgu s kumuláciou Cu v bazálnych gangliách. Zdá sa, že progresiu hepatálneho postihnutia pri WD nedeterminuje špecifický genotyp, lebo genotypovo-fenotypová korelácia sa na veľkom počte pacientov nepotvrdila [3–5]. Procházková pozorovala miernejší stupeň hepatálneho postihnutia a neskorší nástup klinických príznakov u nosičov mutácie H1069Q, ale naši probandi s rovnakým genotypom sa líšili klinickou symptomatológiou či stupňom laboratórnej aktivity choroby. Možno špekulovať, že vek začiatku a priebeh WD determinuje kombinácia vnútorných či vonkajších faktorov, rozdielne mutácie a penetrácia či diétne zvyklosti. Z našej kohorty s malým počtom pacientom mali dve deti cirhózu pečene už v čase diagnostiky WD a ďalší traja pacienti podstúpili transplantáciu pečene ešte v detskom veku [20]. Poľskí autori pozorovali progresívne pečeňové zlyhanie u 16,08 % detí [3]. Z analýzy nášho súboru vyplýva, že prítomnosť patologických výsledkov z vyšetrovaných skórovacích markerov (Kayserov-Fleisherov prstenec, neurologická symptomatológia, znížená hodnota ceruloplazmínu, obsah Cu v sušine, mutačná analýza) je dostatočná na definitívne potvrdenie WD. U deviatich pacientov sa súčasne vyskytovali tri priekazné patologické nálezy: znížený ceruloplazmín, pozitívne genetické vyšetrenie a patologická hodnota Cu v sušine. Sedem detí malo okrem zníženého ceruloplazmínu pozitívnu genetiku (tab. 4). Cielený genetický skríning sme indikovali u súrodencov zo štyroch rodín pacientov s WD. Limitáciou našej štúdie je retrospektívny charakter a relatívne malý počet pacientov. Na druhej strane, veľkosť slovenskej kohorty demograficky „kopíruje“ populáciu našej krajiny v porovnaní s Českom a Poľskom. So zreteľom na tento aspekt sú výsledky prvej slovenskej kohorty relevantným príspevkom ku klinickému priebehu WD u detí v stredoeurópskom regióne.
Záver
WD je pre pestré klinické príznaky, nešpecifické biochemické markery či veľký počet mutácií otvorenou výzvou pediatrov. Potenciálnych jedincov treba aktívne a systematicky vyhľadávať. Ochorenie je poddiagnostikované a „tichí“ pacienti sú dlhodobo sledovaní v špecializovaných ambulanciách bez adekvátnej liečby. Po WD treba aktívne pátrať v neurologických, psychiatrických, psychologických ambulanciách. Veľkým prínosom k včasnému odhaleniu najmä asymptomatických jedincov je cielený screening prvostupňových príbuzných. Tlak na medicínsku komunitu je o to väčší, že WD patrí k mála zriedkavým ochoreniam, ktoré majú účinnú terapiu. Medikamentózna liečba, penicilamínové a zinkové preparáty či trientine dihydrochloride efektívne zabraňujú progresii ochorenia a postihnutým jedincom umožňujú dobrú kvalitu života.
Ďakujeme doc. MUDr. D. Procházkovej, PhD. a Oddeleniu lekárskej genetiky MU v Brne za rozšírenú genetickú diagnostiku u slovenských detí a kolektívu Hepatologického laboratória VFN v Prahe za vyšetrenie obsahu Cu v sušine u slovenských pacientov.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Doručeno/Submitted: 23. 10. 2017
Přijato/Accepted: 4. 12. 2017
MUDr. Jana Kosnáčová
Detská klinika LF UK a DFNsP
Limbová 1
833 40 Bratislava
Slovenská republika
kosnacova@centrum.sk
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Tanner S. Disorders of copper metabolism. In: Kelly DA. Diseases of the liver and biliary system in children. Oxford: Blackwell Science 2004: 328.
2. The Human Gene Mutation Database. Gene Symbol. [online]. Available from: http: //www.hgmd.cf.ac.uk/ac/gene.php?gene=ATP7B.
3. Naorniakowska M, Dądalski M, Kamińska D et al. Clinical presentations of Wilson disease among Polish children. Dev Period Med 2016; 20 (3): 216–221.
4. Møller LB, Horn N, Jeppesen TD et al. Clinical presentation and mutations in Danish patients with Wilson disease. Eur J Hum Genet 2011; 19 (9): 935–941. doi: 10.1038/ejhg.2011.80.
5. Vrabelova S, Letocha O, Borsky M et al. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab 2005; 86 (1–2): 277–285.
6. Mareček Z. Současné možnosti diagnostiky a léčby Wilsonovy choroby. Prakt Lék 2007; 87 (1): 17–22.
7. Procházková D. Diagnostika a terapie dědičných poruch metabolizmu. Wilsonova choroba. Brno: Masarykova univerzita 2004.
8. Schilsky ML. Wilson disease. Current status and the future. Biochemie 2009; 91: 1278–1281. doi: 10.1016/j.biochi.2009. 07.012.
9. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47 (6): 2089–2111. doi: 10.1002/hep.22261.
10. Coffey AJ, Durkie M, Hague S. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013; 136 (Pt 5): 1476–1487. doi: 10.1093/brain/awt035.
11. Litwin T, Gromadzka G, Członkowska A. Gender differences in Wilson’s disease. J Neurol Sci 2012; 312 (1–2): 31–35. doi: 10.1016/j.jns.2011.08.028.
12. European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol 2012; 56 (3): 671–685. doi: 10.1016/j.jhep.2011.11. 007.
13. Sócio S, Ferreira AR, Fagundes ED et al. Wilson’s disease in children and adolescents: diagnosis and treatment. Rev Paul Pediatr 2010; 28 (2): 134–140.
14. Kumar MK, Kumar V, Singh PK et al. Wilson’s disease with neurological presentation, without hepatic involvement in two siblings. J Clin Diagn Res 2013; 7 (7): 1476–1478. doi: 10.7860/JCDR/2013/ 5974.3188.
15. Caca K, Ferenci P, Kühn HJ et al. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol 2001; 35 (5): 575–581.
16. Ferenci P, Caca K, Loudianos G at al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003; 23 (3): 139–142.
17. Sternlieb I. Perspectives on Wilson’s disease. Hepatology 1990; 12 (5): 1234–1239.
18. Kosnáčová J, Podracká Ľ, Hornová J. Výsledky transplantácií pečene u slovenských detí. Gastroent Hepatol 2015; 69 (6): 541–546. doi: 10.14735/amgh2015541.
19. Procházková D, Konečná P, Vrábelová S et al. Význam molekulárně-genetického vyšetření pro diagnostiku Wilsonove choroby. Čes-slov Pediat 2005; 60 (4): 188–199.
20. Dhawan A, Taylor RM, Cheeseman P et al. Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transpl 2005; 11 (4): 441–448.