Vrozené cholestatické syndromy = Inherited cholestatic syndromes
Soňa Režnáková1, Milan Jirsa Orcid.org 2, Martin Hřebíček Orcid.org , Libor Vítek Orcid.org
+ Pracoviště
Souhrn
Režnáková S, Jirsa M, Hřebíček M, Vítek L. Vrozené cholestatické syndromy
Práce uvádí přehled současných poznatků o geneticky podmíněných poruchách sekrece žluči, mezi které patří progresivní familiární intrahepatální cholestáza, benigní rekurentní intrahepatální cholestáza, některé formy intrahepatální těhotenské cholestázy a poruchy syntézy žlučových kyselin. V závěru je uveden stručný seznam dalších vzácných nozologických jednotek. Nejčastější vrozenou cholestázou je deficit kanalikulární pumpy pro lecitin ABCB4/MDR3, který zahrnuje široké spektrum projevů od progresivní familiární intrahepatální cholestázy 3. typu po cholestázu indukovanou orálními kontraceptivy.
Klíčová slova: intrahepatální cholestáza - benigní rekurentní intrahepatální cholestáza - progresivní familiární intrahepatální cholestáza - intrahepatální těhotenská cholestáza - poruchy syntézy žlučových kyselin.
Summary
Režnáková S, Jirsa M, Hřebíček M, Vítek L. Inherited cholestatic syndromes
An overview of the recent knowledge of hereditary disorders of bile secretion - progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis, intrahepatic cholestasis of pregnancy and disorders of bile salt synthesis - is presented. A brief list of other rare disorders is added at the end of the article. Defect in the canalicular lecithin pump ABCB4/MDR3 that involves a broad spectrum of diseases from progressive familial intrahepatic cholestasis type 3 to contraceptive pill-induced cholestasis represents the most frequent hereditary disorder of bile secretion.
Key words: intrahepatic cholestasis - progressive familial intrahepatic cholestasis - benign recurrent intrahepatic cholestasis - intrahepatic cholestasis of pregnancy - disorders of bile salt synthesis.
ÚVOD
Cholestáza je definována jako porucha sekrece nebo odtoku žluči v důsledku strukturálních nebo biochemických abnormalit jaterních buněk a žlučovodů(1). Intrahepatální cholestatické syndromy (ICS) zahrnují onemocnění jater a žlučových cest, jejichž příčina tkví v hepatocytech (tzv. kanalikulární typ cholestázy) nebo v drobných intrahepatálních žlučovodech (tzv. duktální typ cholestázy).
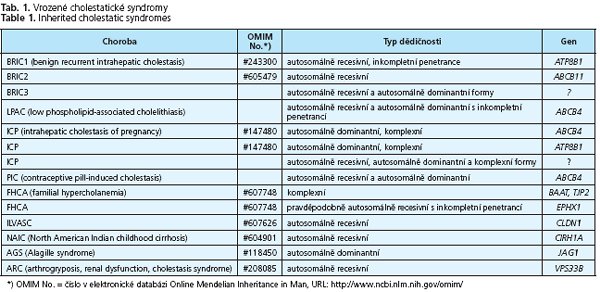
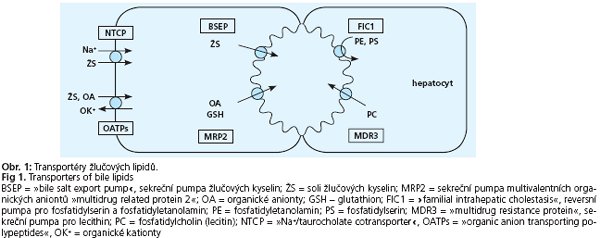
Onemocnění lze klasifikovat podle několika kritérií. Podle etiologie rozeznáváme ICS doprovázející záněty jater, ICS vyvolané autoimunitními mechanismy, ICS vyvolané léky a hormony, ICS v důsledku infiltrace jaterní tkáně nádorovými buňkami či materiálem při střádavých nemocích. Svou roli v etiopatognezi hraje také sepse a celková parenterální výživa, známá je cholestáza v těhotenství, v pediatrii pak vrozené cholestatické syndromy na podkladě genetického defektu transportu žlučových lipidů do žluči (obr. 1.). Podle věku, kdy se tato onemocnění manifestují, se cholestatické syndromy dělí na ICS vyskytující se převážně v dospělosti a na ICS manifestující se již v raném věku. Předmětem následujícího sdělení je stručný přehled nejčastějších vrozených cholestatických syndromů (tab. 1.).
Progresivní familiární intrahepatální cholestáza
Progresivní familiární intrahepatální cholestáza (PFIC) je souhrnný název pro geneticky heterogenní skupinu autosomálně recesivně dědičných nemocí dětského věku podmíněných mutacemi v genech kódujících kanalikulární proteiny odpovědné za transport žlučových kyselin a fosfolipidů z hepatocytů do žluči(2).
Klinicky se PFIC obvykle manifestuje v 1. roce života žloutenkou, těžkým pruritem, hepatosplenomegalií, steatoreou, anorexií, retardací růstu a psychického vývoje. Dále bývají přítomny symptomy deficitu vitaminů rozpustných v tucích: koagulopatie, osteopenie, neuromuskulární poruchy(3). Bez adekvátní léčby nemoc progreduje do jaterní fibrózy až cirhózy a končí obvykle smrtí v důsledku jaterního selhání v první či vzácněji ve druhé dekádě. Konzervativní léčba zahrnuje podávání kyseliny ursodeoxycholové, simvastatinu, cholestyraminu a rifampicinu a bývá obvykle bez většího efektu(2). Chirurgická léčba - ileální bypass či nověji parciální drenáž žluči cholecystostomií(4) u prvních dvou typů PFIC - oddálí potřebu jaterní transplantace, která je v současné době jedinou účinnou terapeutickou možností.
Progresivní familiární intrahepatální cholestáza 1. typu (PFIC1), dříve Bylerova nemoc, je podmíněna defektem v genu kódujícím ATPázu ATP8B1/FIC1 (Familial Intrahepatic Cholestasis 1) lokalizovaném na 18q21(5). Protein ATP8B1 je exprimován v apikální membráně hepatocytů, cholangiocytů, enterocytů s maximem v ileu, buněk pankreatu a v mnohých dalších tkaních(6). V hepatocytech se ATP8B1 podílí na sekreci žlučových kyselin z hepatocytů do žluči, přesný molekulární mechanismus však není objasněn. Kromě výše popsaných příznaků má PFIC1 i symptomy extrahepatální - vodnatý průjem, zhoršení sluchu, pankreatitidy. Ty přetrvávají i po transplantaci jater. Laboratorně dominuje nález cholestázy s nízkou sérovou koncentrací GMT. Koncentrace cholesterolu je normální. Sérová koncentrace primárních žlučových kyselin je zvýšena. Hodnoty jaterních aminotransferáz jsou iniciálně v referenčním rozmezí, s progresí choroby stoupají až na desetinásobek normálních hodnot(1). Histologický nález závisí na pokročilosti nemoci - různě těžký stupeň jaterní fibrózy až cirhózy, cholestáza. V elektronové mikroskopii je typický nález »Bylerovy« hrubě granulární žluči ve žlučových kanalikulech. Diagnostický význam má imunohistochemický průkaz absence proteinu ATP8B1 v kanalikulární membráně. Vzhledem k tomu, že neexistuje protilátka proti ATP8B1 vhodná pro použití v parafinových řezech, je nutno provádět imunohistologické vyšetření v čerstvě zmrazeném vzorku jaterní tkáně.
Progresivní familiární intrahepatální cholestáza 2. typu (PFIC2) je způsobena defektem v genu ABCB11/BSEP (bile salt export pump) kódujícím ATP-dependentní transportér žlučových kyselin. Gen ABCB11 se nachází na 2q24. Protein BSEP je exprimován pouze v kanalikulární membráně hepatocytů(7,8). Klinický průběh i laboratorní nález jsou podobné PFIC1, extrahepatální příznaky však nejsou přítomny. Výjimkou je časná elevace aktivit aminotransferáz. V jaterní biopsii je typický obraz velkobuněčné hepatitidy.
Elektronmikroskopicky je intrakanalikulární žluč amorfní nebo filamentózní. Spolehlivé odlišení PFIC1 a PFIC2 je možné pouze imunohistologickým a/nebo molekulárněgenetickým vyšetřením. Imunohistologický průkaz deficitu BSEP je možno provést v parafinových, formolem fixovaných řezech. Neléčená PFIC2 obvykle vykazuje rychlejší progresi jaterní fibrózy, nicméně po transplantaci jater dochází k ústupu všech příznaků onemocnění. Možnou komplikací PFIC2 je vznik hepatobiliárních malignit, které nebyly zatím popsány u PFIC1. typu.
Progresivní familiární intrahepatální cholestáza 3. typu (PFIC3) je podmíněna mutací v genu ABCB4/MDR3 (multidrug resistance 3) lokalizovaném na 7q21(9). Protein ABCB4 je exprimován v kanalikulární membráně hepatocytů a odpovídá za sekreci fosfolipidů z hepatocytů. U pacientů s defektem ABCB4 pozorujeme absenci fosfatidylcholinu ve žluči, přičemž koncentrace žlučových kyselin je normální. Důsledkem je porucha tvorby smíšených micel a poškození jaterního parenchymu vyvolané detergentním účinkem volných žlučových kyselin na apikální membránu hepatocytů a buněk výstelky žlučových kanálků.
Laboratorně je přítomna cholestáza provázená zvýšenou sérovou aktivitou GMT, což umožňuje odlišení od předchozích dvou typů PFIC. V jaterní histologii dominuje těžká fibróza až cirhóza s portální zánětlivou infiltrací a duktulární proliferace(9,10). Imunohistologicky lze obvykle prokázat nepřítomnost proteinu ABCB4 v játrech, vyšetření je možno provést i v parafinových řezech.
Benigní rekurentní intrahepatální cholestáza
Benigní rekurentní intrahepatální cholestáza (BRIC) je soubor vrozených autosomálně recesivních, geneticky heterogenních onemocnění projevujících se intermitentními atakami cholestázy(11). Genetickým podkladem nemoci jsou mutace v genech ATP8B1 a ABCB11 (tedy genech zapříčiňujících také syndromy PFIC1 a PFIC2, viz výše) a velmi pravděpodobně i ve třetím dosud neidentifikovaném genu(12). Analogicky ke klasifikaci PFIC můžeme i BRIC rozdělit na BRIC1 a BRIC2(13).
Zatímco mutace nalezené u PFIC jsou obvykle lokalizovány v oblastech kódujících konzervované funkční domény proteinu a závažně poškozují funkci těchto proteinů, u BRIC se jedná o mutace v méně konzervovaných oblastech s částečným dopadem na funkci proteinu(13). Choroba se typicky manifestuje před 2. dekádou, věk první manifestace je ale velmi variabilní. Rovněž variabilní je i doba trvání cholestatické epizody (několik dnů až několik měsíců) a její intenzita. Spouštěcím faktorem ataky může být interkurentní infekce či těhotenství. V klinickém obrazu dominuje ikterus, pruritus, únava, anorexie a steatorea.
V době trvání ataky nacházíme laboratorně elevaci bilirubinu a žlučových kyselin v séru, aktivita GMT a sérová koncentrace cholesterolu je normální. Koncentrace aminotransferáz jsou normální nebo jen lehce zvýšené. V mezidobí se všechny biochemické parametry vracejí do normálního rozmezí. V jaterní biopsii provedené v době trvání cholestatické ataky dominuje obraz cholestázy, struktura jaterního parenchymu je zcela normální, nejsou známky jaterní fibrózy či cirhózy. V asymptomatickém období je histologický obraz zcela normální. Průběh onemocnění je typicky benigní, zhoršuje ale kvalitu života, invalidizuje pacienta, intenzivní pruritus může vést v extrémních případech k suicidálním pokusům. Terapie cholestyraminem a ursodeoxycholovou kyselinou může zmírnit subjektivní potíže, vliv terapie na prevenci vzniku další ataky je sporný(11). Nedávno byly publikovány případy pacientů, u nichž se původně rekurentně probíhající choroba změnila po několika letech v progredující postižení s rozvojem jaterní cirhózy(11). Proto v současné době chápeme BRIC a PFIC jako dva extrémy kontinuálního spektra fenotypů jedné nemoci(1,14).
Intrahepatální těhotenská cholestáza
Intrahepatální těhotenská cholestáza (ICP), postihující v České republice kolem 1 % gravidit, je charakterizovaná vznikem cholestázy obvykle v 3. trimestru gravidity bez známek jaterní nemoci v předchorobí. Typickým příznakem je pruritus, který se s pokračující graviditou stupňuje. ICP způsobuje fetální distres, předčasný porod nezralého plodu nebo i intrauterinní odumření plodu(15). V laboratorním nálezu je zvýšená koncentrace sérových žlučových kyselin a aktivita jaterních aminotransferáz, v těžších případech dochází i k elevaci bilirubinu a cca u 10-15 % postižených žen i k vzestupu aktivity GMT. Léčba kyselinou ursodeoxycholovou má na průběh gravidity příznivý vliv. Jaterní testy se normalizují spontánně bezprostředně po porodu(15). U některých žen s ICP byly nalezeny heterozygotní mutace v genu MDR3(16-19). Předpokládá se, že za normálních okolností poloviční exprese proteinu stačí na zabezpečení normální žlučové sekrece, avšak při extrémní hormonální zátěži organismu dojde k manifestaci poruchy. Stejným mechanismem se vysvětluje i cholestatický syndrom indukovaný perorálními kontraceptivy. Byly popsány rovněž asociace ICP s mutacemi v ATP8B1(20).
Adultní formy poruchy biliární sekrece fosfolipidů
Mutace, které nevedou k úplné ztrátě schopnosti vylučovat do žluči fosfolipidy, mohou kromě těhotenské intrahepatální cholestázy vyvolat i různě rychle progredující chronické jaterní onemocnění s mírnou trvalou či intermitentní cholestázou. Histologicky bývá na počátku onemocnění přítomna portální fibróza s minimální zánětlivou celulizací, která může progredovat až do obrazu jaterní cirhózy. Dalšími projevy částečného deficitu ABCB4/MDR3 jsou familiární cholesterolová cholelitiáza(21,22) a intrahepatální cholestáza indukovaná hormony (např. kontraceptivy) (23). Vznik cholelitiázy je výsledkem nedostatečné transportní kapacity žluči obsahující nízké množství fosfolipidů pro cholesterol. Význam adultních forem deficitu genu ABCB4 dokresluje i naše vlastní zkušenost: z 25 probandů vyšetřených s podezřením na dědičně podmíněnou cholestázu bylo u 12 vysloveno podezření na deficit MDR3, který jsme u 7 z nich potvrdili mutační analýzou.
Vrozené defekty syntézy žlučových kyselin
Vrozené defekty syntézy žlučových kyselin se mohou rovněž manifestovat cholestázou. Základem je vyšetření koncentrace celkových žlučových kyselin v séru. Normální koncentrace žlučových kyselin u pacienta s cholestázou vylučuje PFIC a budí podezření na defekt syntézy žlučových kyselin. Podmínkou interpretovatelnosti nálezu je přerušení léčby kyselinou ursodeoxycholovou minimálně 2 týdny před odběrem vzorku krve či moči. Ke stanovení koncentrace žlučových kyselin, jejich prekurzorů a metabolitů se používá FAB-MS (fast atom bombardment-mass spectroscopy) pro stanovení v moči a GC-MS (gas chromatography-mass spectroscopy) pro stanovení v séru(24). Normální nebo nízká koncentrace primárních žlučových kyselin společně s nízkou aktivitou GMT svědčí pro poruchu tvorby žlučových kyselin, naopak zvýšené koncentrace spolu s nízkou aktivitou GMT pozorujeme u PFIC1, PFIC2 a některých dalších vzácných cholestatických nemocí. Terapie spočívá v substituci chybějících primárních žlučových kyselin.
Ostatní vzácné dědičné cholestázy
Familiární hypercholanémie (FHCA): první typ FHCA je autosomálně recesivně dědičné onemocnění podmíněné mutací v genu pro BAAT (bile acid CoA:amino acid N-acyltransferase) (25). Enzym BAAT katalyzuje konjugaci žlučových kyselin s aminokyselinou taurinem či glycinem. Nemoc se klinicky projevuje pruritem a příznaky malabsorpce tuků, v laboratorním nálezu dominuje extrémně zvýšená koncentrace primárních žlučových kyselin v séru. Příčinou zvýšení koncentrace žlučových kyselin je zpětná difúze nekonjugovaných žlučových kyselin do plazmy přes bazolaterální membránu hepatocytů, nebo» nekonjugované žlučové kyseliny nejsou vylučovány do žluči. Druhý typ FHCA je způsoben homozygotní mutací v genu pro tight junction protein 2 (TJP2) (25). Těsná spojení (tight junctions) hepatocytů a buněk výstelky žlučových cest oddělují žluč od plazmy v Disseho prostoru. Defekt v TJP2 zvyšuje permeabilitu těchto spojení pro žluč, což způsobuje únik žlučových kyselin do plazmy. Výsledkem je vysoká plazmatická koncentrace žlučových kyselin s pruritem na jedné a malabsorpcí tuků na druhé straně.
ILVASC: autosomálně recesivní syndrom zahrnující ichtyózu, leukocytární vakuoly, alopecii a sklerozující cholangitidu je podmíněn mutacemi v genu CLDN1 kódujícím další z »tight junction« proteinů. Abnormality desmosomů vedou ke zvýšení jejich propustnosti v játrech (26) .
NAIC (North American Indian childhood cirrhosis): autosomálně recesivní cholestatické onemocnění s rozvojem fibrózy a následně cirhózy v dětském věku způsobené mutací v genu CIRH1A (27) kódujícím nukleolární protein cirhin, jehož funkce zatím není objasněna.
Alagillův syndrom: autosomálně dominantně podmíněná vývojová porucha charakterizována multiorgánovým postižením s těžkou cholestázou při vrozené chybné diferenciaci intrahepatálních žlučovodů na podkladě mutace v genu JAG1 kódujícím mezibuněčný signální protein Jagged 1.
ARC syndrom: artrogrypóza, renální dysfunkce, cholestáza a trombocytopatie asociovaná s mutací v genu VPS33B kódujícím protein regulující fúzi nitrobuněčných membránově vázaných organel(28).
ZÁVĚR
V našem přehledu jsme shrnuli nové poznatky o vrozených cholestatických syndromech. Problematika je komplikovaná a odráží složitost molekulárních pochodů uvnitř jaterní buňky, které se uplatňují v biliární sekreci žlučových lipidů. Během posledních deseti letech došlo v této oblasti k velkému rozvoji poznatků na úrovni patogeneze a diagnostiky. Je nutno zdůraznit, že základní biochemické a molekulárněgenetické metody pro diagnostiku vrozených cholestatických syndromů jsou v České republice k dispozici na pracovištích autorů a tento fakt by měl vést k lepšímu záchytu a přesnější diagnostice nemocných. V blízké budoucnosti lze očekávat, že s dalším rozvojem našeho poznání se i klinická medicína dočká účinných kauzálních léků pro terapii vrozených ICS.
Literatura
- 1. van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis 2001; 21: 535-544.
- 2. Kotalova R, Cebecauerova D, Knisely AS, et al. Progressive familial intrahepatic cholestasis - manifestations and diagnosis in infancy. Cesk Pediatr 2006; 61: 200-206.
- 3. Thompson R, Strautnieks S. BSEP: function and role in progressive familial intrahepatic cholestasis. Semin Liver Dis 2001; 21: 545-550.
- 4. Kalicinski PJ, Ismail H, Jankowska I, et al. Surgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg 2003; 13: 307-311.
- 5. Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998; 18: 219-224.
- 6. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004; 40: 27-38.
- 7. Gerloff T, Stieger B, Hagenbuch B, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 1998; 273: 10046-10050.
- 8. Jansen PL, Muller M. The molecular genetics of familial intrahepatic cholestasis. Gut 2000; 47: 1-5.
- 9. de Vree JM, Jacquemin E, Sturm E, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci USA 1998; 95: 282-287.
- 10. Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001; 120: 1448-1458.
- 11. van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol 2002; 36: 439-443.
- 12. Floreani A, Molaro M, Mottes M, et al. Autosomal dominant benign recurrent intrahepatic cholestasis (BRIC) unlinked to 18q21 and 2q24. Am J Med Genet 2000; 95: 450-453.
- 13. van Mil SW, van der Woerd WL, van der Brugge G, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology 2004; 127: 379-384.
- 14. Carlton VE, Pawlikowska L, Bull LN. Molecular basis of intrahepatic cholestasis. Ann Med 2004; 36: 606-617.
- 15. Lammert F, Marschall HU, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy: molecular pathogenesis, diagnosis and management. J Hepatol 2000; 33: 1012-1021.
- 16. Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet 2000; 9: 1209-1217.
- 17. Gendrot C, Bacq Y, Brechot MC, Lansac J, Andres C. A second heterozygous MDR3 nonsense mutation associated with intrahepatic cholestasis of pregnancy. J Med Genet 2003; 40: e32.
- 18. Lucena JF, Herrero JI, Quiroga J, et al. A multidrug resistance 3 gene mutation causing cholelithiasis, cholestasis of pregnancy, and adulthood biliary cirrhosis. Gastroenterology 2003; 124: 1037-1042.
- 19. Floreani A, Carderi I, Paternoster D, et al. Intrahepatic cholestasis of pregnancy: three novel MDR3 gene mutations. Aliment Pharmacol Ther 2006; 23: 1649-1653.
- 20. Mullenbach R, Linton KJ, Wiltshire S, et al. ABCB4 gene sequence variation in women with intrahepatic cholestasis of pregnancy. J Med Genet 2003; 40: e70.
- 21. Rosmorduc O, Hermelin B, Poupon R. Mdr3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001; 120: 1459-1467.
- 22. Rosmorduc O, Hermelin B, Boelle PY, et al ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003; 125: 452-459.
- 23. Ganne-Carrie N, Baussan C, Grando V, et al. Progressive familial intrahepatic cholestasis type 3 revealed by oral contraceptive pills. J Hepatol 2003; 38: 693-694.
- 24. Bove KE, Heubi JE, Balistreri WF, Setchell KD. Bile acid synthetic defects and liver disease: a comprehensive review. Pediatr Dev Pathol 2004; 7: 315-334.
- 25. Carlton VE, Harris BZ, Puffenberger EG, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet 2003; 34: 91-96.
- 26. Baala L, Hadj-Rabia S, Hamel-Teillac D, et al. Homozygosity mapping of a locus for a novel syndromic ichthyosis to chromosome 3q27-q28. J Invest Dermatol 2002; 119: 70-76.
- 27. Chagnon P, Michaud J, Mitchell G, et al. A missense mutation (R565W) in cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am J Hum Genet 2002; 71: 1443-1449.
- 28. Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet 2004; 36: 400-404.
Finanční zdroje: M.J. a M.H. byli podpořeni grantem IGA MZČR č. NR9079-3/2006.
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené