Subkutánně podávaný vedolizumab pro ulcerózní kolitidu a Crohnovu chorobu v klinickém programu VISIBLE
Úvod
Idiopatické střevní záněty (IBD – inflammatory bowel diseases) jsou skupinou chronických zánětlivých autoimunitně podmíněných onemocnění zažívacího ústrojí, které zahrnují Crohnovu chorobu (CN) a ulcerózní kolitidu (UC). Jedná se o civilizační nemoci, které postihují osoby nejčastěji ve druhém až čtvrtém decenniu a jejichž etiologie je stále ne zcela jasná. V léčbě IBD se uplatňuje medikamentózní terapie aminosalicyláty, glukokortikoidy, imunosupresivy, antibiotiky a enterální výživou. V případech, kdy není zřejmá odpověď na podávanou léčbu, připadá v úvahu chirurgický výkon či biologická terapie. Monoklonální protilátky proti tumor nekrotizujícímu faktoru alfa (anti-TNFα) jsou základními pilíři biologické léčby IBD a jsou k dispozici pro léčbu CN a UC v České republice již několik let [1].
Vedolizumab
Novější variantu biologické léčby představují protilátky proti integrinovým receptorům na povrchu cirkulujících aktivovaných B a T lymfocytů. Tyto integriny se vyvazují na specifické ligandy, které jsou exprimovány na povrchu endoteliálních buněk v trávicí trubici. Nazýváme je adheziny (MAdCAM-1 a VCAM-1). Vedolizumab je humanizovaná monoklonální protilátka IgG1 pro integrinový receptor α4β7. Provedený klinický výzkum GEMINI I a II prokázal vysokou efektivitu a bezpečnost intravenózně podávaného vedolizumabu v léčbě nemocných se středně až vysoce aktivní UC a CN. Výhodou se jeví také vysoká účinnost u nemocných, kteří selhali na anti-TNFα léčbě, selektivní účinek na trávicí trakt a příznivý bezpečností profil léčiva [2,3].
Subkutánně podávaný vedolizumab
Vedolizumab je první biologickou léčbou, která je v Evropě schválena jak pro intravenózní, tak pro subkutánní podání v udržovacím režimu středně těžké až těžké UC a CN. Tato unikátní možnost volby typu aplikace v udržovací léčbě jistě povede k vyšší compliance léčby u pacientů a nám lékařům umožní lepší a snadnější rozhodování, kdy nebudeme limitováni jen jedním typem možného podání.
Efektivitu léčby a bezpečnost sledoval klinický program VISIBLE 1 u UC, VISIBLE 2 u CN a následně v dlouhodobé extenzi s open label subkutánním podáním VISIBLE OLE.
VISIBLE 1 (příloha 1) je dvojitě slepá, dvojitě maskovaná a placebem kontrolovaná randomizovaná studie fáze III u pacientů se středně těžkou až těžkou aktivní UC, kteří nedostatečně reagovali na předchozí medikamentózní léčbu kortikosteroidy a/nebo imunosupresivy a/nebo anti-TNFα terapii.
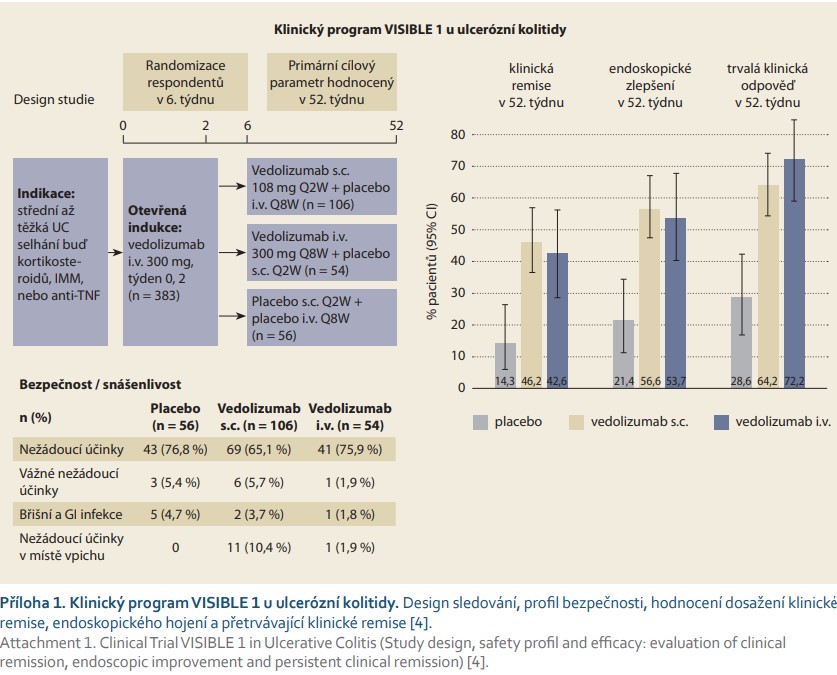
V úvodu studie byly všem pacientům podány dvě intravenózní infuze v dávce 300 mg v týdnu 0 a 2, a pokud byla zaznamenána odpověď na léčbu v týdnu 6, byli pacienti randomizováni do tří větví. První s vedolizumabem i.v. po 8 týdnech a placebem po 2 týdnech, s vedolizumabem s.c. po 2 týdnech a placebem po 8 týdnech, nebo s placebem (jak s.c. po 2 týdnech, tak i.v. po 8 týdnech). Primárním cílem bylo dosažení klinické remise v týdnu 52, sekundárními cíli pak endoskopické zlepšení (slizniční hojení) a přetrvávající klinická remise v týdnu 52. Klinická remise byla definována jako kompletní Mayo skóre ≤ 2, slizniční hojení jako Mayo endoskopické subskóre ≤ 1 a přetrvávající klinická remise jako remise v týdnu 6 a 52. Klinické remise dosáhlo 46,2 % pacientů ve větvi s vedolizumabem s.c. (p < 0,001), 42,6 % pacientů ve větvi s vedolizumabem i.v. a ve větvi placeba 14,3 %. Pokud se podíváme na dosažení klinické remise u pacientů naivních na anti-TNFα, pak ve skupině s s.c. vedolizumabem dosáhlo klinické remise 53,7 % (p < 0,001) pacientů, u i.v. léčby vedolizumabem 53,1 %. Oproti tomu ve skupině pacientů selhaných na anti-TNFα byla klinická remise u 33,3 % (p = 0,023) v s.c. skupině, 27,3 % u i.v. podávané léčby. Endoskopického zlepšení v týdnu 52 dosáhlo v s.c. vedolizumabové větvi 56,6 % pacientů (p < 0,001), 53,7 % pacientů v i.v. vedolizumabové větvi a 21,4 % ve větvi s placebem. Hodnotu přetrvávající klinické remise dosáhlo větší množství pacientů v s.c. vedolizumabové větvi (15,1 %) oproti placebové větvi (5,4 %), ale výsledek nebyl statisticky signifikantní (p = 0,076). Parametru přetrvávající klinické odpovědi v týdnu 52 dosáhlo 64,2 % pacientů v s.c. vedolizumabové větvi (p < 0,001), 72,2 % ve větvi i.v. vedolizumabu a v placebové větvi 28,6 % pacientů.
Pacientům, kteří v týdnu 6 neodpověděli na léčbu, byla podána i.v. infuze v týdnu 6 a 14, a ti, kteří dosáhli klinické odpovědi, pokračovali v programu VISIBLE OLE. Průměrný věk pacientů zařazených do VISIBLE 1 je 39,7 let, délka trvání UC 7,9 let, 28 % pacientů před vstupem do studie selhalo na léčbě anti-TNFα a 23,7 % užívalo před zařazením do studie kortikosteoroidy [4].
Studie VISIBLE 1 u pacientů s těžkou až středně těžkou UC prokázala, že subkutánně podávaný vedolizumab je efektivní a dobře tolerovaný. Dosažení klinické remise v týdnu 52 bylo výrazně vyšší než u placeba stejně jako dosažení endoskopického zhojení a pokračující klinické remise. Dále výsledky studie VISIBLE 1 naznačují, že subkutánní léková forma vedolizumabu je v udržovací léčbě srovnatelně účinná a srovnatelně bezpečná jako forma intravenózní.
VISIBLE 2 (příloha 2) je dvojitě zaslepená, placebem kontrolovaná, randomizovaná studie fáze III u pacientů se středně těžkou až těžkou CN. Celkem 644 (z tohoto počtu 39 % anti-TNFα naivních) pacientům byla podána indukce stejně jako u VISIBLE 1. Všichni dostali dvě i.v. infuze vedolizumabu v dávce 300 mg, a pokud byla zaznamenána klinická odpověď (hodnoceno jako pokles CDAI > 70 bodů) v týdnu 6, byli randomizováni do větve s aktivním s.c. vedolizumabem (108 mg s.c. v intervalu à 2 týdny) nebo do větve s placebem. Primárního endpointu, tj. klinické remise, dosáhlo 48 % pacientů s vedolizumabem a 34,3 % pacientů s placebem (p = 0,008). Sekundárním cílem byla přetrvávající klinická odpověď, dosažení remise bez kortikoidů (corticosteroid free clinical remission) a dosažení klinické remise u pacientů dosud biologicky naivních. Přetrvávající klinická odpověď byla nastolena u 52 % pacientů s vedolizumabem a u 44,8 % pacientů s placebem (p = 0,167). Remise bez kortikoidů byla zaznamenána u 45,3 % pacientů s vedolizumabem a u 15,2 % s placebem, klinické remise dosáhlo 48,6 % pacientů naivních na anti-TNFα ve srovnání s 42,9 % u placebové skupiny. Lokální reakce v místě vpichu byla hlášena u < 3 % pacientů, závažné nežádoucí účinky byly v obou ramenech < 5 % a protilátky proti vedolizumabu byly zaznamenány u tří pacientů (2,5 %) [5].
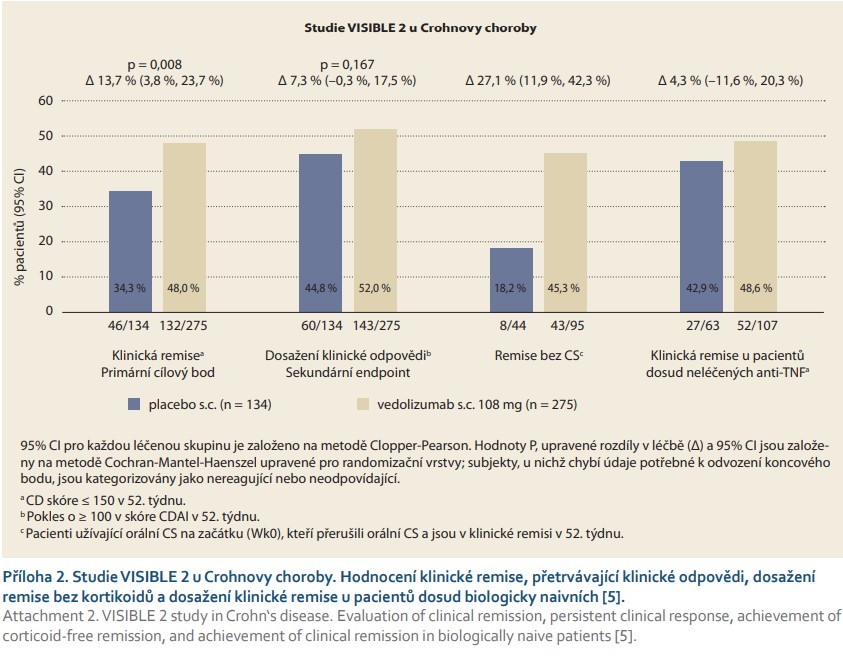
Studie VISIBLE 2 u pacientů s CN prokázala vyšší dosažení klinické remise ve skupině subkutánně podaného vedolizumabu ve srovnání s placebem. Nežádoucí účinky byly malé a bezpečností profil je konzistentní a srovnatelný s intravenózní aplikací.
VISIBLE OLE je pokračující, dlouhodobá, otevřená, multicentrická studie fáze III hodnotící bezpečnost a snášenlivost vedolizumabu podávaného subkutánně ve studiích VISIBLE 1 i 2. Design studie spočívá v podání s.c. vedolizumabu u pacientů, kteří buď dokončili 52. týden, nebo museli předčasně studii ukončit (s možností navýšení frekvence podání z 2 týdnů na jednotýdenní interval). Nemocní s nedostatečnou odpovědí v úvodu obou studií v týdnu 6 dostali další i.v. infuzi, a pokud byla zaznamenána zřejmá odpověď v týdnu 14, pokračovali opět do open label extenze – VISIBLE OLE. Průběžná analýza studie VISIBLE OLE byla zveřejněna na kongresu UEG Week Virtual 2020 a týkala se výsledků bezpečnosti a účinnosti subkutánního vedolizumabu v udržovací léčbě nemocných se středně těžkou až těžkou UC. Účastníci, kteří ve VISIBLE 1 dokončili 52 týdnů udržovací léčby nebo kteří dosáhli klinické odpovědi ve 14. týdnu po třetí infuzi vedolizumabu i.v. v 6. týdnu, dostávali v otevřeném pokračování vedolizumab s.c. 108 mg každé dva týdny. Během následujících dvou let prodlouženého sledování nebyly pozorovány žádné nové nepopsané nežádoucí účinky. Mírné nebo středně závažné nežádoucí účinky se vyskytly u 69 % pacientů, nejčastěji byly hlášeny exacerbace onemocnění (18 %), nazofaryngitida (11 %), infekce horních cest dýchacích (9 %) a anémie (7 %). Reakce v místě vpichu byly hlášeny u 4,5 % pacientů. Závažné nežádoucí účinky se vyskytly u 14 % pacientů, nebyl však zaznamenán ani jeden případ progresivní multifokální leukoencefalopatie nebo úmrtí. Dlouhodobá účinnost byla hodnocena jako dlouhodobé udržení klinické remise (definované jako parciální Mayo skóre ≤ 2 bez individuálního podskóre > 1) a klinické remise bez kortikosteroidů. U randomizovaných dokončivších byla až do 108. týdne sledování udržena míra dosažení klinické remise (v 6. týdnu 71 % a v 108. týdnu 68,9 %) i klinické remise bez kortikosteroidů (v 52. týdnu 78,3 % a v 108. týdnu 70 %). U nerandomizovaných respondentů ve 14. týdnu byla míra dosažení klinické remise 62,6 % ve 14. týdnu a 33,3 % ve 110. týdnu. Míra dosažení klinické remise bez kortikosteroidů činila 24,5 % v 54. týdnu a 25 % ve 110. týdnu sledování [5].
U pacientů, u nichž byla intenzifikována léčba na jednotýdenní interval pro známky selhání, dosáhlo 27,1 % opětovné klinické remise po 16 týdnech intenzifikace a 10,8 % jich setrvalo v klinické remisi i v týdnu 48 [6,7]. Zatím však nejsou k dispozici kompletní výsledky studie.
Závěr
Na základě výše uvedených výsledků je zřejmé, že subkutánní léková forma vedolizumabu je v udržovací léčbě srovnatelně účinná a bezpečná jako forma intravenózní. Obě lékové formy jsou v udržovací léčbě z hlediska klinického významu a použití zaměnitelné.
MUDr. Barbora Pipek
Centrum péče o zažívací trakt
Nemocnice AGEL Ostrava-Vítkovice a. s.
Zalužanského 1192/15
703 84 Ostrava-Vítkovice
barbora.pipek@vtn.agel.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Lukáš M. Perspektivy biologické léčby u idiopatických střevních zánětů. Gastroent Hepatol 2014; 68 (3): 225–229.
2. Bortlík M. Vedolizumab – nová antiintegrinová protilátka s vysokou gastrointestinální selektivitou. Gastroent Hepatol 2014; 68 (6): 481–484. doi: 10.14735/amgh2014481.
3. Sandborn WJ, Feagan BG, Rutgeerts P et al. Vedolizumab as induction and maintenance therapy for Crohn‘s disease. N Engl J Med 2013; 369 (8): 711–721. doi: 10.1056/NEJMoa1215739.
4. Sandborn WJ, Baert F, Danese S et al. Efficacy and safety of vedolizumab subcutaneous formulation in a randomized trial of patients with ulcerative colitis. Gastroenterology 2020; 158 (3): 562–572.e12. doi: 10.1053/j.gastro.2019.08.027.
5. Vermeire S, Sandborn W, Baert F et al. OP23 Efficacy and safety of vedolizumab SC in patients with moderately to severely active Crohn’s disease: results of the VISIBLE 2 study. J Crohns Colitis 2020; 14 (Suppl 1): S020–S021. doi: 10.1093/ecco-jcc/jjz203.022.
6. Loftus EV jr, Sandborn WJ, Wolf D et al. P499 Efficacy and safety of 2 or 3 vedolizumab intravenous infusions as induction therapy for ulcerative colitis and Crohn’s disease: results from VISIBLE 1 and 2. J Crohns Colitis 2019; 13 (Suppl 1): S361–S362. doi: 10.1093/ecco-jcc/ jjy222.623.
7. Sandborn WJ, Wolf D, D’haens G et al. P510 Dose escalation of subcutaneous vedolizumab in patients with ulcerative colitis: a post hoc analysis of the VISIBLE trial data. J Crohns Colitis 2020; 14 (Suppl 1): S442–S443. doi: 10.1093/ecco-jcc/jjz203.639.